Introduction
Plasma cell neoplasms (PCN) is a group of mature B-cell disorders that result from an abnormal generation of cloned plasma cells that produce monoclonal, heavy-chain, class-switched immunoglobulin referred to as paraprotein or M-protein. According to the WHO classification in 2017, this class of neoplasms includes non-IgM monoclonal gammopathy of undetermined significance (MGUS), plasmacytoma, monoclonal immunoglobulin deposition diseases, and plasma cell myeloma (PCM), and plasma cell neoplasms with the associated paraneoplastic syndrome (see Table. WHO Classification of Plasma Cell Neoplasms).[1][2]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiology of plasma cell neoplasms is currently unknown. However, chronic antigenic stimulation from infections, chronic disease, radiation, or specific toxic substances has been proposed. Studies have shown that there is increased risk due to exposure to specific substances in occupations such as cosmetologists, farmers, and chronic laxative abusers. A hereditary factor is possible; in non-immunoglobulin (Ig)-M monoclonal gammopathy of undetermined significance, an increased prevalence of the disease has been found in families with a lymphoproliferative disorder [3].
Epidemiology
PCM is the most common disease entity in this category because it is symptomatic and well-studied. PCM comprises about 1% of malignant tumors, 10% to 15% of hematopoietic neoplasms, and causes up to 20% of the deaths in the hematologic malignancies category. PCM is a disease of older adults (eg, 50 or older), with a median age at diagnosis of 66 years. There is an increased risk in males and African Americans. Non-Ig-M monoclonal gammopathy of undetermined significance is a PCM precursor disease and presents in up to 4% of plasma cell neoplasms [3].
Pathophysiology
The pathophysiology of plasma cell neoplasms is related to the monoclonal plasma cell proliferation and the disease entity. The abnormal plasma cell clone can cause lytic lesions in the bone, inhibit normal hematopoiesis, and lead to deterioration of renal function through the deposition of abnormal immunoglobulin. Amyloid light-chain (AL) amyloidosis pathogenesis is due to amyloid deposition in tissues; AL is composed of intact immunoglobulin light chains or less commonly, heavy chains that are produced by the monoclonal plasma cells and then taken over and discharged by macrophages into the extracellular matrix.
History and Physical
The clinical presentation of plasma cell neoplasms largely depends on the specific disease. The first step in approaching any suspected plasma cell neoplasm patient is to obtain a thorough history, clinical examination, and laboratory tests to verify whether the patient’s diagnosis is PCM or a premalignant stage of PCM, such as MGUS or smoldering PCM, which does not require therapy and may be easily misdiagnosed as PCM. MGUS patients are usually asymptomatic with a risk of progression of approximately 1%. MGUS has been reported to progress to PCM, solitary plasmacytoma, or amyloidosis [4].
PCM is the main disease entity in plasma cell neoplasms and presents variably, with symptoms ranging from asymptomatic to aggressive. The diagnosis is based on the assessment of pathological, radiological, and clinical features (table 2). Symptoms of PCM include symptoms related to bone marrow infiltration (fatigue and pallor, bleeding, and infections) and symptoms related to monoclonal gammopathy, often referred to as CRAB symptoms (hypercalcemia, renal insufficiency, anemia, and bone lesions). Extramedullary involvement leads to masses/organomegaly with lymphadenopathy, hepatomegaly, and splenomegaly being the most common organs involved. The manifestation of advanced diseases such as fatigue, generalized weakness, weight loss, and fever are possible presenting symptoms [3].
PCM has the following 3 variants:
- Asymptomatic (smoldering) plasma cell myeloma shows classic M protein and bone marrow findings with no symptoms.
- Non-secretory myeloma shows an absence of an M-protein; however, elevated serum free light chains and/or an abnormal free light chain ratio are detectable.
- Plasma cell leukemia (PCL) shows extensive and diffuse involvement by clonal plasma cells (greater than 60%) and association with extramedullary tissue infiltration as well as increased incidence of CRAB symptoms.[5]
Plasmacytoma is generally classified into solitary bone plasmacytoma (SBP) and extramedullary (extraosseous) plasmacytoma (EMP). SBP refers to the involvement of the cortical bone with monoclonal plasma cells. The most common bone sites involved include the vertebrae, ribs, and skull. SBP presents as localized bony pain and may have soft tissue extension. SBP shows progression to PCM or multiple plasmacytoma in up to 70% of cases, therefore close observation and follow up is crucial. SBP can be confused with early PCM, therefore complete and careful assessment to exclude PCM is crucial, including clinical examination, tests to assess for M-protein, bone marrow aspirate/biopsy with no evidence of clonal plasma cells, and normal skeletal survey except for the primary solitary lesion identified. Solitary plasmacytoma with minimal bone marrow involvement refers to a variant of SBP where clonal plasma cells of less than 10% have been identified in the bone marrow biopsy [5].
EMP is localized plasma cell neoplasms that arise in tissues other than bone. The distinction between lymphomas that exhibit extreme plasma cell differentiation and EMP may be difficult. Examples of lymphomas with prominent plasmacytic differentiation that need to be differentiated from EMP include extranodal marginal zone (MALT) lymphoma and extranodal marginal zone lymphoma (MZL). EMP occurs commonly in the upper respiratory tract (80%; sinuses, naso- and oropharynx); symptoms are generally related to the tumor mass including rhinorrhea, epistaxis, and nasal obstruction. The second most common site of EMP involvement is the gastrointestinal tract (5%); symptoms include loss of appetite, bleeding, abdominal discomfort, or obstruction. Other organs that can be involved include the lung, lymph nodes, bladder, CNS, breast, thyroid, testis, parotid, and skin [5].
Monoclonal immunoglobulin deposition diseases include primary amyloidosis (AL) and light and heavy chain deposition diseases. Both of these disorders are characterized by the deposition of aberrant immunoglobulin in soft and visceral tissue, leading to organ dysfunction; the major difference between the two disease entities is that AL stains positive with Congo red stain. Primary AL is more common than light and heavy chain deposition diseases, very rare disease entities. AL is caused by a plasma cell neoplasm that secretes intact or fragments of abnormal immunoglobulin light chains, or less commonly, heavy chains, which deposit in various tissues. This disease entity is associated with PCM in approximately 20% of patients, therefore thorough assessment is crucial. Sites of involvement include subcutaneous fat, kidney, heart, liver, gastrointestinal tract, peripheral nerves, and bone marrow. Clinical symptoms are extremely variable and depend on the organs involved. For instance, heart involvement may present as congestive heart failure, kidney involvement may present as nephrotic syndrome, gastrointestinal involvement may present as malabsorption, and liver involvement may present as hepatomegaly. As the clinical symptoms vary, clinical suspicion of the disease should always be kept in mind.
Plasma cell neoplasms with associated paraneoplastic syndrome include two disease entities: POEMS syndrome and TEMPI syndrome (provisional disease entity). POEMS syndrome is an osteosclerotic myeloma that is characterized by fibrosis and osteosclerotic changes in bone trabeculae and lymph node changes similar to the plasma cell variant of Castleman disease. This rare syndrome's symptoms include polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes (POEMS). The characteristic lesion in osteosclerotic plasmacytoma may be single or multiple. Most patients do not present with all the manifestations of POEMS syndrome. The most common presenting symptom is chronic, progressive polyneuropathy.
Evaluation
Investigations for plasma cell neoplasms include the following tests to identify the disease entity. Initial laboratory work set for plasma cell neoplasms includes a complete blood count (CBC), peripheral blood smear (PB), serum calcium and creatinine, serum protein electrophoresis (SPEP) and immunofixation, urine protein electrophoresis (UPEP) and immunofixation, serum free light chain (FLC) assay and quantitative immunoglobulins. If the tests yield a high suspicion for PCM, the following laboratory tests are then added: Bone marrow biopsy/bone marrow aspirate, routine urine analysis, and 24-hr urine collection for electrophoresis and immunofixation, serum albumin, Lactate dehydrogenase (LDH), beta-2-microglobulin and C-reactive protein (CRP). In PCM, the PB smear may show anemia, plasmacytosis, Rouleaux formation, or leucoerythroblastic reaction. Bone marrow biopsy/bone marrow aspirate is a crucial test in the diagnosis of PCN. Plasma cell identification, enumeration, and clonality are all assessed on the biopsy specimen. Immunohistochemical staining for CD38, CD138, Ig kappa, and lambda light chains is used to identify plasma cells in the bone marrow. Staining for the aberrant CD56 and KIT may also be useful. However, they are not used routinely in clinical practice. Clonal plasma cells greater than 10% are considered positive for PCM, while less than 10% of clonal bone marrow plasma cells are considered positive for non-Ig M MGUS. Morphologically, the plasma cells can be immature, pleomorphic, or mature. In AL bone marrow biopsy/bone marrow aspirate is extremely variable and can show no pathological findings, extensive amyloid deposition, PCM findings, or lymphoplasmacytic lymphoma. Immunohistochemical techniques using anti-amyloid fibril antibodies to kappa and lambda are useful in distinguishing primary and secondary amyloidosis (AA) in some cases [4][1].
Immunophenotyping utilizing flow cytometry is another useful diagnostic tool that usually complements and confirms the morphological assessment of the biopsy. The plasma cells are CD38 +ve, CD138 (bright) +ve, CD45 –ve, and CD19 –ve. Aberrant antigens such as CD56, CD200, KIT, CD28, CD52, and others are commonly identified. It is important to note that no association between immunophenotyping and prognosis has been found. An M protein is measured in urine and serum and is one of the diagnostic criteria for the diagnosis of PCM (greater than 30g/L) and aids in differentiating this entity from non-Ig M MGUS. Lytic bone lesions on imaging are estimated to be present in about 70% of cases of PCM patients and are considered one of the diagnostic criteria of diagnosis. Imaging modalities that are used include XR, PET-CT, and MRI [1][2].
Treatment / Management
The treatment and management of plasma cell neoplasms depend on the disease entity. Non-IgM MGUS is an asymptomatic disease with a low risk of progression, so it is usually managed through close follow-up. SBP and EMP are treated with localized radiotherapy. Although standard treatment of plasmacytomas is absent, plasmacytomas are highly sensitive to radiotherapy; therefore, local radiation therapy is effective [4][6]. PCM treatment depends on risk stratification. Following risk stratification, all patients need to be assessed to determine their eligibility for autologous hematopoietic cell transplantation (HCT). When compared with chemotherapy alone, intensified chemotherapy followed by HCT has been shown to improve both the disease-free survival and the overall survival. For high and intermediate-risk groups, induction chemotherapy followed by HCT and maintenance therapy is recommended. If a patient is not a candidate for high-dose chemotherapy and autologous HCT, the only treatment option is chemotherapy alone. Triple chemotherapeutic regimens such as bortezomib, lenalidomide, dexamethasone (VRd); bortezomib, cyclophosphamide, dexamethasone (VCd); and bortezomib, melphalan, prednisone (VMP) are all possible options. Initial chemotherapy should be continued until the patient reaches a plateau phase (12 to 18 months) in which the M-protein is stabilized and the disease shows no progression. The preferred method to assess the response to therapy is the measurement of monoclonal (M) protein in serum and urine. Amyloidosis and light and heavy chain deposition diseases are generally managed similarly to PCM. Eligibility for HCT should be determined, and chemotherapy alone should be used only in HCT-ineligible patients. Overall, plasma cell neoplasm management requires multidisciplinary care and prompt risk stratification and treatment for high-risk diseases [4][6][2].
Differential Diagnosis
- Amyloidosis
- Immunoglobin-related amyloidosis
- Light chain-associated renal disorders
- Light chain deposition disease
- Monoclonal gammopathies of uncertain origin
- Multiple myeloma
- Plasmacytoma
Prognosis
The prognosis of plasma cell neoplasms depends on the disease entity. PCM prognosis depends on multiple factors including disease stage, genetics, host factors (age, performance status, co-morbidities), response to therapy, disease biology, and others. The Durie and Salmon (OS) staging system applies clinical parameters to predict myeloma cell tumor burden and prognosis and is generally classified as low, intermediate, and high. The International staging system (ISS) for PCM is based on the serum beta-2 microglobulin and albumin levels and has been shown to predict survival. Genetics is used to classify PCM prognosis as standard, intermediate, and high-risk. The high-risk group includes t (14; 16), t (14; 20), and deletion 17p13 and gene expression profiling (high risk). The intermediate risk group includes deletion 13, t (4; 14), and hypodiploidy. The favorable risk group includes hyperdiploidy, t (11; 14) and t (6; 14). Hyperdiploidy is generally defined as chromosomes greater than 48 and less than 75.[2][4]
Non-Ig M MGUS is generally not considered a neoplastic process with a risk of progression of approximately 1%. Progression of non-Ig M MGUS to PCM, solitary plasmacytoma, or amyloidosis has been reported. Factors influencing the progression of MGUS include the size and type of M proteins. Immunoglobulins less than 5 g/L and greater than 25 g/L are markers of low and high risk of progression. Non-Ig M MGUS rarely presents with abnormal karyotypes. EMP has a better prognosis than PCM and SBP. Early PCM, may be misdiagnosed as EMP. A thorough and complete assessment of the patients, as well as a long follow-up, is recommended. SBP shows progression to PCM in up to 60% of patients. EMP has an indolent course with recurrences occurring in up to 25% of patients, distant metastasis is relatively uncommon, and progression to PCM is infrequent and occurs in 20% of cases.[2]
Monoclonal immunoglobulin deposition disorders have a poor prognosis, with cardiac and hepatic failure and infection being the major causes of death. The median survival for patients with primary amyloidosis is approximately 2 years; while patients with both amyloidosis and PCM have even a shorter survival expectancy. Recently, the survival of these diseases has improved, especially when no cardiac involvement is present. On the other hand, POEMS syndrome has a favorable overall prognosis compared to other plasma cell neoplasms, with an expected median overall survival of 14.7 years. Reports of cure have been reported with localized radiation therapy.
Enhancing Healthcare Team Outcomes
PCM is managed by an interprofessional team that includes an oncology nurse. PCM treatment depends on risk stratification. Following risk stratification, all patients need to be assessed to determine their eligibility for autologous hematopoietic cell transplantation (HCT). When compared with chemotherapy alone, intensified chemotherapy followed by HCT has been shown to improve both the disease-free survival and the overall survival. For high and intermediate-risk groups, induction chemotherapy followed by HCT and maintenance therapy is recommended. If a patient is not a candidate for high-dose chemotherapy and autologous HCT, the only treatment option is chemotherapy alone.
Media
(Click Image to Enlarge)
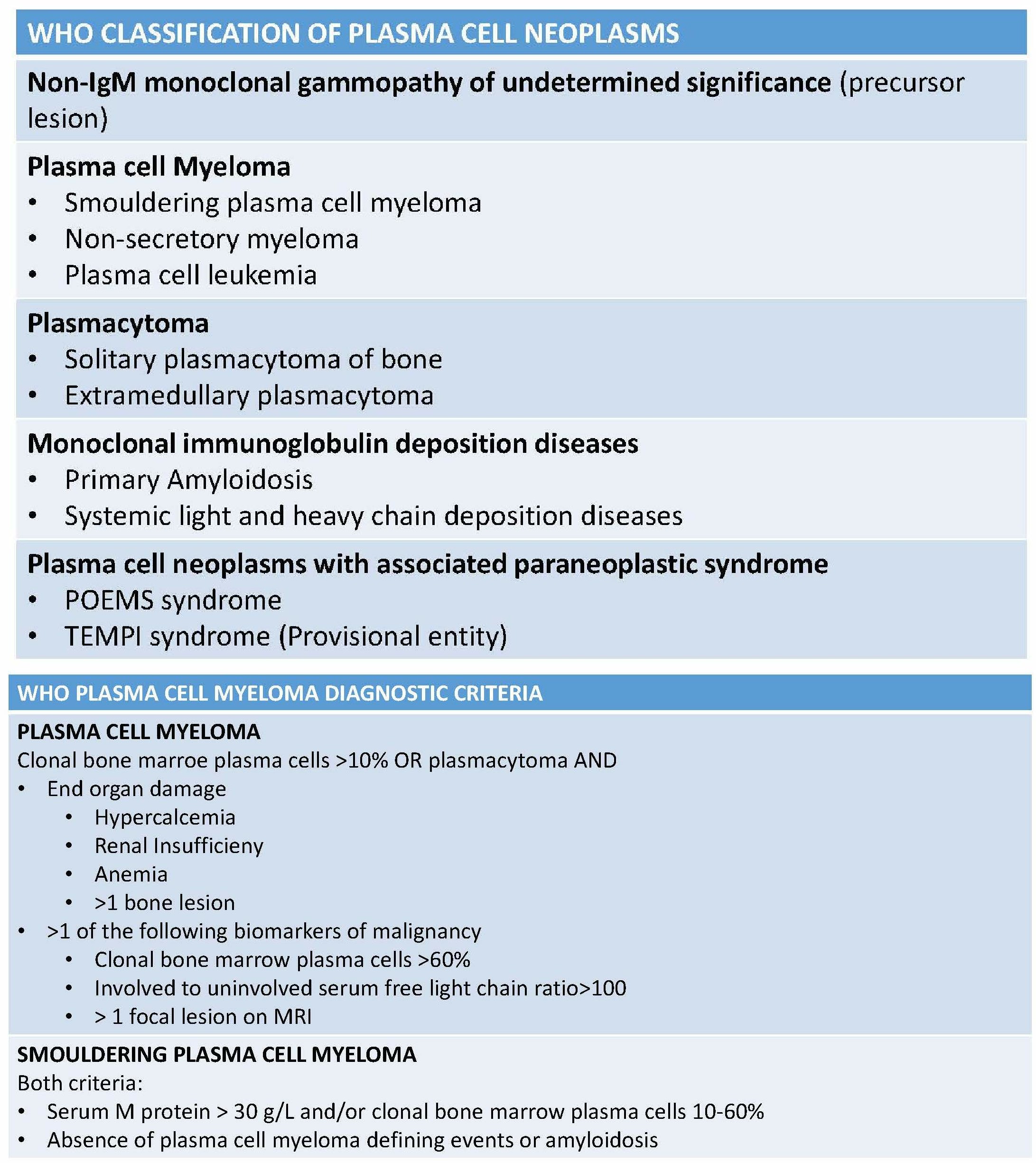
WHO Classification of Plasma Cell Neoplasms
H Kaseb, MD
References
Kyle RA, Gertz MA, Witzig TE, Lust JA, Lacy MQ, Dispenzieri A, Fonseca R, Rajkumar SV, Offord JR, Larson DR, Plevak ME, Therneau TM, Greipp PR. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clinic proceedings. 2003 Jan:78(1):21-33 [PubMed PMID: 12528874]
Level 2 (mid-level) evidenceRajkumar SV. Multiple myeloma: 2012 update on diagnosis, risk-stratification, and management. American journal of hematology. 2012 Jan:87(1):78-88. doi: 10.1002/ajh.22237. Epub [PubMed PMID: 22180161]
Kyle RA, Rajkumar SV. Multiple myeloma. The New England journal of medicine. 2004 Oct 28:351(18):1860-73 [PubMed PMID: 15509819]
Multiple myeloma in young patients: clinical presentation and treatment approach., Bladé J,Kyle RA,, Leukemia & lymphoma, 1998 Aug [PubMed PMID: 9711912]
Caers J, Paiva B, Zamagni E, Leleu X, Bladé J, Kristinsson SY, Touzeau C, Abildgaard N, Terpos E, Heusschen R, Ocio E, Delforge M, Sezer O, Beksac M, Ludwig H, Merlini G, Moreau P, Zweegman S, Engelhardt M, Rosiñol L. Diagnosis, treatment, and response assessment in solitary plasmacytoma: updated recommendations from a European Expert Panel. Journal of hematology & oncology. 2018 Jan 16:11(1):10. doi: 10.1186/s13045-017-0549-1. Epub 2018 Jan 16 [PubMed PMID: 29338789]
Rajkumar SV. Treatment of multiple myeloma. Nature reviews. Clinical oncology. 2011 Apr 26:8(8):479-91. doi: 10.1038/nrclinonc.2011.63. Epub 2011 Apr 26 [PubMed PMID: 21522124]