Introduction
Pulmonary vascular resistance (PVR) is the resistance against blood flow from the 4 pulmonary veins of the lung to the left atrium. It is most commonly modeled using a modification of Ohm’s law (see Image. Pulmonary Vascular Resistance Derived from Ohm's Law). Input pressure represents the mean pulmonary arterial pressure (15 mm Hg). The output pressure represents the pulmonary venous pressure, equivalent to the pulmonary capillary wedge or left atrial pressure (5 to 6 mm Hg). Total blood flow represents the cardiac output (5 to 6 L/min). A normal value for pulmonary vascular resistance using conventional units is 0.25 to 1.6 mmHg·min/l. Pulmonary vascular resistance is also represented in units of dynes/sec/cm-5 (normal = 37dynes/sec/cm-5 to 250 dynes/sec/cm-5).[1]
Poiseuille’s law has been used to model PVR (see Image. Poiseuille's Law Modeling Pulmonary Vascular Resistance). In this equation, l represents the length of the tube or vessel, r its radius, and n the fluid's viscosity. Poiseuille’s law clarifies the impact of the radius on resistance. For example, a 50% reduction in radius increases the resistance 16-fold. However, Ohm’s and Poiseuille’s laws are approximations of PVR. Both equations assume that blood flow is constant and linear, but it is pulsatile and laminar. Pulmonary blood vessels are dynamic, multi-layered tissues that expand to accommodate increased flow. Additionally, the non-homogenous quality of blood makes it difficult to ascertain a single value for viscosity. Blood viscosity varies with shear rate.[2][3]
The pressure drop from the pulmonary veins to the left atrium is approximately 10 mm Hg compared to a 100 mm Hg pressure gradient in the systemic circulation. Therefore, PVR is one-tenth of the resistance of systemic circulation. Low PVR maximizes the distribution of blood to the peripheral alveoli and facilitates gas exchange. Low resistance also enables the pulmonary system to pump the total cardiac output at low pressures.
Most of the total vascular resistance and distribution of blood flow in the pulmonary circuit is due to the cross-sectional area of capillaries rather than veins, venules, or arteries. However, pulmonary resistance is approximately equally divided between arteries, capillaries, and veins. Because resistance increases in the capillaries, the largest drop in pulmonary pressure occurs, and to a lesser extent, in the small pulmonary arteries. In the systemic circulation, the largest pressure drop occurs in the arterioles.[1]
Mechanism
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Mechanism
Multiple mechanisms regulate and contribute to pulmonary vascular resistance. Broad categories include pulmonary vascular pressure, lung volume, gravity, smooth muscle tonicity, and alveolar hypoxia.[4]
Pulmonary Intravascular Pressure
As cardiac input increases, for example, during exercise, the pulmonic circulation must adapt to accommodate this increased forward flow. Therefore, pulmonary intravascular pressure and pulmonary vascular resistance are inversely related. Experiments have shown that increasing the pulmonary arterial pressure while holding the left atrial pressure constant decreases pulmonary vascular resistance.
This decrease occurs via 2 mechanisms: capillary recruitment and capillary distension.
The first mechanism is capillary recruitment. Some pulmonary capillaries are partially or entirely closed at baseline, impeding blood flow. Capillary recruitment refers to opening closed capillaries during states of increased blood flow. Flow distribution over a greater cross-sectional surface area reduces the overall vascular resistance. Recruitment usually occurs in zone 1 of the lung (apices), where the capillary pressures are the lowest.
Capillary distension is the second mechanism and involves widening the capillaries to accommodate increased blood flow. The Ovular vessels become more circular, the predominant mechanism for maintaining low PVR at higher pulmonary arterial pressures.[1]
Lung Volumes
Alveolar pressures and volumes influence pulmonary vascular resistance. The effect of lung volume depends on the type of vessel. Extra-alveolar vessels run through the lung parenchyma. These vessels have smooth muscle and elastic tissue, which reduces vessel circumference by counteracting distension. As the lung expands, the diameter of these vessels increases via radial traction of the vessel walls. Therefore, vascular resistance is low at large lung volumes. During low lung volumes, there is high resistance due to the unopposed action of vessel elasticity.
Critical opening pressure represents the air pressure facilitating blood flow through extra-alveolar capillaries. This concept applies when modeling vascular resistance in a collapsed lung with low volume.
Alveolar capillaries include capillaries and vessels in the corner of the alveolar walls. Their transmural pressure determines the amount of lung distension.
Alveolar pressure is highest in zone 1 (near the apices) and lowest in zone 3 (near the bases). During inspiration, alveolar pressure rises, compressing the surrounding alveolar capillaries. With increased right heart return on inspiration, stretching and thinning of the alveolar walls reduces capillary caliber and increases PVR. PVR is highest at total lung capacity (TLC), high at residual volume (RV), and lowest at functional residual capacity (FRC) (see Image. Standard Lung Volumes and Capacities).[5]
Gravity
Three zones of the lung are typically identified (see Image. Normal PA Chest X-Ray Delineated by Lung Zones). PVR is greatest at zone 1 since the elevated alveolar pressure increases the inward transmural pressure on the alveolar-capillary membrane. The capillary becomes collapsible, and the resistance increases. PVR is lowest at zone 3, where the arterial pressure is higher than the alveolar pressure, causing an increased outward transmural pressure and vessel caliber.[6]
Alveolar Hypoxia
Hypoxia within the alveoli induces vasoconstriction within the lung vasculature. This homeostatic mechanism allows the lungs to shunt blood to more oxygenated lung segments, increasing the ventilation/perfusion (V/Q) ratio and improving oxygen delivery throughout the body.
This mechanism is important when the lungs are exposed to disruptive processes, such as consolidation (eg, pneumonia) or blockage within the vasculature (eg, pulmonary emboli), leading to physiological compensation. The theory is that this response begins at the molecular level when a mitochondrial sensor utilizes redox coupling reactions to alter the elasticity of pulmonary artery smooth muscle cells (PASMC).
The redox reactions lead to the depolarization of PASMC via activation of voltage-gated calcium channels and inhibition of potassium channels, causing decreased elasticity within the arterioles of hypoxic lung segments. If hypoxia is sustained, alternative pathways are activated (eg, rho kinase), and the chemokine release (eg, hypoxia-inducible factor (HIF)-1 alpha) leads to vasoconstrictive effects and vasculature remodeling.[7]
Smooth Muscle Tonicity
Generally, the pulmonary circulation has a low vascular tone due to pulmonary vessels having proportionately less smooth muscle than vessels of similar diameter in other organs. Compared to systemic vessels, the smooth muscle tissue in pulmonary vessels is distributed less evenly in the tunica intima. The pulmonary veins are also more compliant than systemic arteries due to the lack of tissue around small vessels, reduced elastin and collagen fibers, and reduced smooth muscle content. This phenomenon is demonstrated by the pressure gradient between the right and left ventricles.[2][8]
Pulmonary arteries are both elastic and muscular. These arteries contain smooth muscle within the tunica media surrounded by internal and external elastic laminae. These include the pulmonary artery trunk, main branches, and extra-alveolar vessels. Larger, peri-bronchial arteries are more muscular (>2mm). Peri-bronchial arteries lie within the lung lobules. These extra-alveolar arteries control PVR through neural, humoral, or gaseous mediators. As the vessels are smaller, smooth muscle content decreases. The smooth muscle takes on a spiral shape and forms the pulmonary arterioles that supply alveoli and alveolar ducts. Smooth muscle exceeding 5% of the external diameter is considered pathological.
Pulmonary arteries have more smooth muscle than veins and represent vasoactive mediators' constriction sites. Capillaries are devoid of vasomotor control. Factors that cause increased tone and increased PVR include serotonin, epinephrine, norepinephrine, histamine, ATP, adenosine, neurokinin A, endothelin, angiotensin, thromboxane A/prostaglandins/leukotrienes (LTB). Most of these factors act through a G-protein coupled pathway, which activates myosin contraction. Neuronally, pulmonary constriction is under the mediation of the sympathetic nervous system by stimulation of a1 adrenergic receptors.[9]
Factors that decrease smooth muscle tonicity and PVR include acetylcholine and isoproterenol, prostacyclin (PGI), bradykinin, vasopressin, ANP, substance P, VIP, and histamine (during adrenaline response). Most of the factors act through activation of cyclic adenosine 3’,5’ monophosphate (cAMP). cAMP de-phosphorylates myosin and reduces calcium levels, causing relaxation of smooth muscle. Pulmonary endothelial cells cause relaxation by producing nitric oxide (NO).
NO diffuses through smooth muscle cells and activates cyclic guanosine 3’, 5’ monophosphate (cGMP), which causes smooth muscle relaxation by the de-phosphorylation of myosin. Additionally, stimuli from the parasympathetic nervous system via the vagus nerve on M muscarinic receptors in the vasculature cause NO-dependent vasodilation.[9]
Pathophysiology
Disease processes that cause chronic hypoxia increase pulmonary vascular resistance through hypoxic pulmonary vasoconstriction that remodels the vessels of the pulmonary circulation over time. These include pulmonary edema, pulmonary emboli, and cardiovascular disease.
Pulmonary edema causes alveoli to shrink due to increased surface tension, causing hypoxemia and shunting of blood to areas of higher ventilation. Hypoxia induces vasoconstriction, leading to pulmonary hypertension and increased vascular resistance. Peri-bronchial cuffing increases the resistance of extra-alveolar vessels, while alveolar edema compresses and distorts capillaries.
Recruitment and distension of pulmonary capillaries provide a large reserve in the pulmonary circulation. Half of the pulmonary circulation can become obstructed before a detectable rise in pulmonary pressure occurs. While the mechanism of increased PVR secondary to pulmonary embolism is still under research, current understanding suggests that it is caused by serotonin release from platelets.
Cardiovascular disease may also lead to increased pulmonary vascular resistance. Left heart valvular disease, such as mitral stenosis or regurgitation, leads to elevated pressures in the left atrium and, ultimately, the pulmonary veins. Increases in pulmonary capillary pressures over a long period lead to smooth muscle hypertrophy and fibrosis of pulmonary vasculature. These changes, in turn, cause pulmonary arterial hypertension and eventually cor pulmonale.
Clinical Significance
Increased pulmonary vascular resistance is the leading cause of pulmonary hypertension.[10] Furthermore, increased PVR can lead to pulmonary hypertension, leading to increased PVR due to chronic vasoconstriction, vascular remodeling, endothelial thickening, arteriolar smooth muscle hypertrophy, and increased thromboxane and endothelin-1 production.
Pulmonary hypertension is a mean pressure greater than 25 mm Hg and a PVR greater than 3 mmHg·min/l. This measurement is obtained through a right heart catheterization (eg, Swan-Ganz catheters).[11]
Increased PVR may occur secondary to processes that cause hypoxic pulmonary vasoconstriction or neurally mediated vasoconstriction (catecholamine release). These processes include chronic obstructive pulmonary disease, emphysema, pulmonary fibrosis, cystic fibrosis, sleep apnea, lupus, scleroderma, rheumatoid arthritis, and HIV. Vascular obstruction secondary to venous thromboembolism, air embolus, amniotic fluid, and schistosomiasis also causes an increase in pulmonary vascular resistance, leading to pulmonary hypertension. Obliterative processes such as pulmonary vasculitides destroy capillary beds, ultimately increasing PVR in certain areas.[7]
Media
(Click Image to Enlarge)
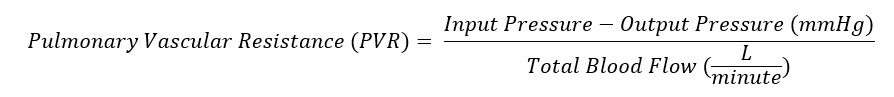
Pulmonary Vascular Resistance Derived from Ohm's Law. Pulmonary vascular resistance derived from Ohm's law, factoring input and output blood pressure in relation to blood flow.
Contributed by Vivek Ashok
(Click Image to Enlarge)
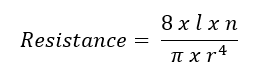
Poiseuille's Law Modeling Pulmonary Vascular Resistance. Resistance is also derived from Poiseuille's Law that can measure blood vessel parameters.
Contributed by Vivek Ashok
(Click Image to Enlarge)
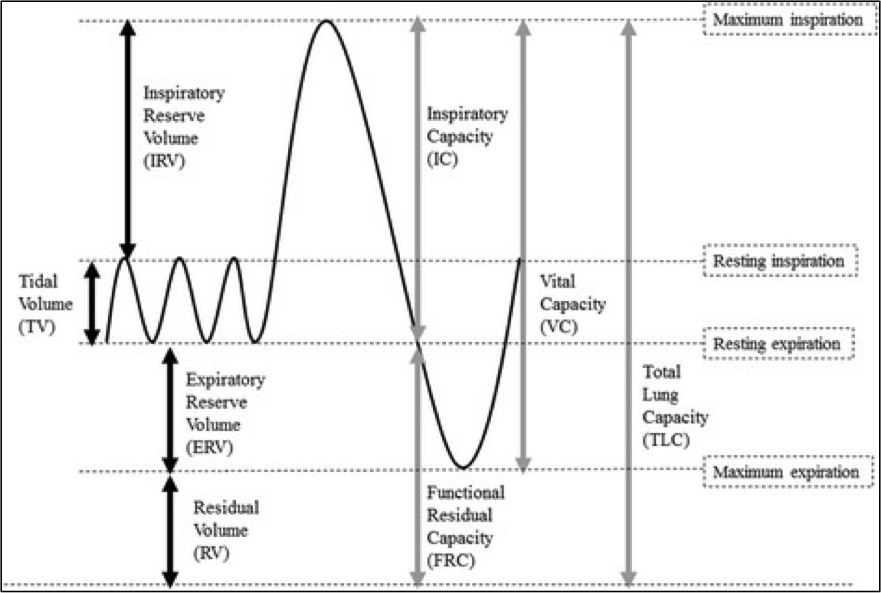
Standard Lung Volumes and Capacities. Standard lung volumes and capacities that may be affected by pulmonary disease.
Contributed by Lutfi MF. The physiological basis and clinical significance of lung volume measurements. Multidisciplinary Respiratory Medicine. 2017;12:3. (CC by 4.0; Creative Commons Attribution 4.0 International License)
(Click Image to Enlarge)
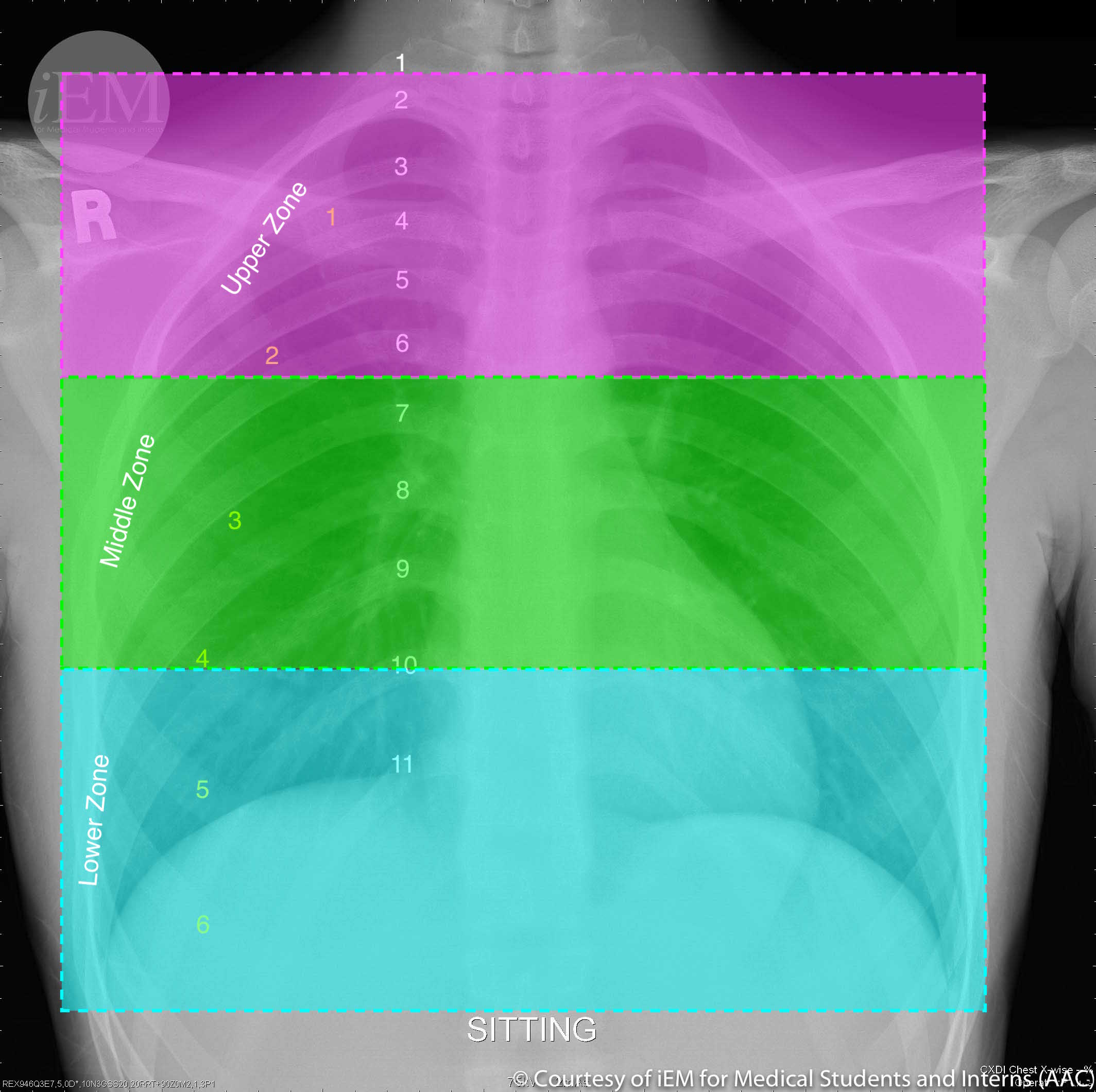
Normal PA Chest X-Ray Delineated by Lung Zones. Normal Chest x-ray showing upper zone: from the apex to 2nd costal cartilage, middle zone: between 2nd and 4th costal cartilage, and lower zone: between 4th and 6th costal cartilage.
iEM for Medical Students and Interns. CC BY-NC-SA 2.0 DEED https://creativecommons.org/licenses/by-nc-sa/2.0/
References
Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circulation research. 2014 Jun 20:115(1):115-30. doi: 10.1161/CIRCRESAHA.115.301146. Epub [PubMed PMID: 24951762]
Ghio S, Schirinzi S, Pica S. Pulmonary arterial compliance: How and why should we measure it? Global cardiology science & practice. 2015:2015(4):58. doi: 10.5339/gcsp.2015.58. Epub 2015 Nov 28 [PubMed PMID: 26779530]
Poullis M. Central and peripheral pulmonary vascular resistance: Implications for who should undergo pulmonary thromboendarterectomy. Medical hypotheses. 2015 Aug:85(2):113-6. doi: 10.1016/j.mehy.2015.03.009. Epub 2015 Mar 10 [PubMed PMID: 25997984]
Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ (Clinical research ed.). 2018 Mar 14:360():j5492. doi: 10.1136/bmj.j5492. Epub 2018 Mar 14 [PubMed PMID: 29540357]
Suresh K, Shimoda LA. Lung Circulation. Comprehensive Physiology. 2016 Mar 15:6(2):897-943. doi: 10.1002/cphy.c140049. Epub 2016 Mar 15 [PubMed PMID: 27065170]
Levitzky MG. Teaching the effects of gravity and intravascular and alveolar pressures on the distribution of pulmonary blood flow using a classic paper by West et al. Advances in physiology education. 2006 Mar:30(1):5-8 [PubMed PMID: 16481601]
Level 3 (low-level) evidenceDunham-Snary KJ, Wu D, Sykes EA, Thakrar A, Parlow LRG, Mewburn JD, Parlow JL, Archer SL. Hypoxic Pulmonary Vasoconstriction: From Molecular Mechanisms to Medicine. Chest. 2017 Jan:151(1):181-192. doi: 10.1016/j.chest.2016.09.001. Epub 2016 Sep 16 [PubMed PMID: 27645688]
Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiological reviews. 2009 Jul:89(3):957-89. doi: 10.1152/physrev.00041.2008. Epub [PubMed PMID: 19584318]
Level 3 (low-level) evidenceGao Y, Chen T, Raj JU. Endothelial and Smooth Muscle Cell Interactions in the Pathobiology of Pulmonary Hypertension. American journal of respiratory cell and molecular biology. 2016 Apr:54(4):451-60. doi: 10.1165/rcmb.2015-0323TR. Epub [PubMed PMID: 26744837]
Dunlap B, Weyer G. Pulmonary Hypertension: Diagnosis and Treatment. American family physician. 2016 Sep 15:94(6):463-9 [PubMed PMID: 27637122]
Kwan WC, Shavelle DM, Laughrun DR. Pulmonary vascular resistance index: Getting the units right and why it matters. Clinical cardiology. 2019 Mar:42(3):334-338. doi: 10.1002/clc.23151. Epub 2019 Feb 27 [PubMed PMID: 30614019]