Introduction
The blood goes from right to left side of the heart through the pulmonary circulation, which is low-resistance, low-pressure circulation. The mean pulmonary vascular pressure is normally between 10 to 18 mm Hg. During exercise, this low-pressure circulation can accommodate high blood flow. The resistance in the pulmonary vasculature depends on the pressure in the pulmonary arteries, left atrial pressure, and cardiac output.
The increase in resistance due to any cause that leads to an abnormal increase in pressures in the pulmonary artery can lead to pulmonary hypertension. Pulmonary hypertension is a persistent increase in the mean pulmonary arterial pressure of more than 25 mm Hg at rest and greater than 30 mm Hg during exercise on cardiac catheterization.
Pulmonary hypertension (PH) can be classified as idiopathic/primary when the cause is unknown. Secondary pulmonary hypertension occurs due to underlying diseases or known risk factors, underlying heart and lung disease being the most common.[1][2]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
WHO classification based on the mechanism or underlying etiology:
- Group 1: Pulmonary arterial hypertension (PAH) can be idiopathic (i.e., primary pulmonary hypertension) or due to the congenital left to right intracardiac shunts, portal hypertension, persistent pulmonary hypertension of the newborn, collagen vascular diseases, HIV infection, drugs, and toxins.
- Group 2: Pulmonary hypertension secondary to left heart disease, valvular heart disease, restrictive cardiomyopathy (pulmonary venous hypertension). The most common causes are mitral valve stenosis and left heart diastolic dysfunction.
- Group 3: Pulmonary hypertension associated with advanced chronic lung disease and environmental hypoxemia. It includes chronic obstructive pulmonary disease (COPD), interstitial lung disease, sleep-disordered breathing, and alveolar hypoventilation disorder.
- Group 4: Pulmonary hypertension due to chronic thrombotic disease, embolic disease, or both
- Group 5: A miscellaneous complex group of disorders leading to PH which includes metabolic disorders, systemic disorders, hematologic diseases, and miscellaneous causes.[3][4][5][6][7][8][9]
- Metabolic disorders such as glycogen storage disease, thyroid disease, Gaucher disease
- Systemic diseases include sarcoidosis, vasculitis, neurofibromatosis type 1.
- Hematologic diseases like myeloproliferative disorders.
- Other causes include end-stage renal disease on dialysis, extrinsic compression of pulmonary vessels, embolization of the tumor [10][11][12][13]
Epidemiology
Idiopathic pulmonary arterial hypertension (IPAH) is a rare form of PH that is more common in females. The incidence of the other groups is similar to the particular underlying diseases.
The prevalence of group 3 pulmonary hypertension (PH) appears to be high in older patients. However, prevalence varies among COPD patients due to genetic polymorphisms, with higher mean pulmonary pressures in COPD patients with LL serotonin transporter gene polymorphism when compared to LS or SS variants.[14][15][16]
Pathophysiology
Pulmonary hypertension irrespective of the cause occurs due to restricted blood flow in a pulmonary artery. The major pathology in Group 1 and Group 4 is obstruction of pulmonary vessels. Some diseases such as sarcoidosis, schistosomiasis, and HIV infection directly affect pulmonary vessels.
Group 1 PH (Pulmonary artery hypertension): This group includes diseases that alter the pulmonary vasculature structurally. The bone morphogenetic protein receptor 2 gene (BMRP2 gene) belongs to the TGF-b family. It normally controls the proliferation of vascular smooth muscle cells. Mutation in BMRP2 gene can result in hypertrophy of smooth muscle along with dysfunction of the endothelium. This causes a decrease in the inner diameter of the lumen of the pulmonary artery that leads to an increase in pulmonary vascular resistance and an increase in right ventricular afterload resulting in high PA pressures.
Group 2 PH (Pulmonary venous hypertension or postcapillary pulmonary hypertension): The left heart disease causes backpressure and increases the hydrostatic pressure in the pulmonary veins which eventually results in increased pressure in pulmonary arteries.
Group 3 PH: Advanced chronic lung conditions lead to lung tissue destruction and a decrease in the capillary area. The common mechanisms that lead to pulmonary hypertension in this group are hypoxic vasoconstriction and vascular remodeling of the pulmonary vasculature.
Hypoxic pulmonary vasoconstriction is a compensatory mechanism that occurs to preserve the ventilation-perfusion matching by limiting the blood flow to hypoxic alveoli. The primary mechanism for vasoconstriction is through mitochondrial signaling and inhibition of potassium channels and pulmonary vascular smooth muscle membrane depolarization, thereby increasing calcium entry in muscle cells. Vascular remodeling also occurs which affects the pulmonary arterioles. These changes occur due to increased expression of endothelin 1, platelet-derived growth factors, vascular endothelial growth factor, and angiotensin 2 and a reduction in nitric oxide synthase.
In Group 4 PH, thromboembolic occlusion or narrowing of pulmonary arteries, proximal and distal both can lead to an increase in backpressure in the pulmonary circulation. This eventually leads to vascular remodeling due to shear stress. The combination of vessel occlusion and vasculopathy increase the resistance and pressure within the pulmonary circulation.[17][9][18][19][20][21]
Group 5 PH is multifactorial.
Histopathology
Pulmonary vasculopathy occurs as a result of shear stress due to chronically elevated pulmonary artery pressures. It includes hypertrophy of the media, intimal fibrosis, and adventitial layer thickening. Concentric lesions of intima can be seen in severe pulmonary hypertension.[22][23]
History and Physical
Dyspnea on exertion, lethargy, and fatigue are the initial symptoms due to the inability to maintain cardiac output, especially during exercise. Exertional dyspnea progresses to dyspnea at rest as the disease advances.
Orthopnea and paroxysmal nocturnal dyspnea can be seen in Group 2 PH patients. Patients can also complain of a nonproductive cough, hemoptysis, hoarseness. Exertional anginal pain, syncope, and peripheral edema can occur due to right ventricular hypertrophy and heart failure as the disease progresses.
The severity is based on New York Heart Association (NYHA) Functional Classification.
Signs include a loud second heart sound pulmonary component, which is the initial physical finding, followed by tricuspid regurgitation murmur, jugular venous distention, hepatomegaly, and edema of lower extremities.
Evaluation
Pulmonary hypertension can be suspected on clinical presentation. However, it is important to rule out other causes as the signs and symptoms of pulmonary hypertension are non-specific. The baseline functional class of the patient can be determined by the 6-minute walk test.
If pulmonary hypertension is suspected, a presumptive diagnosis of pulmonary hypertension can be made with echocardiography, which estimates the systolic pressure of the pulmonary artery and right heart function. Further tests are done to exclude heart and lung diseases that are the common causes of pulmonary hypertension. These tests include ECG, pulmonary function test (PFT) with the diffusing capacity of the lungs for carbon monoxide (DLCO), arterial blood gas (ABG), chest x-ray, and high-resolution computed tomography (HRCT).
ECG shows right ventricular hypertrophy, right atrial enlargement, right axis deviation, increased amplitude of P wave in lead II in all groups. It may show left ventricular hypertrophy in left heart disease.
The chest radiograph may show signs of left heart disease such as pulmonary vascular congestion and pleural effusion. Increased right ventricular size and cardiomegaly in all patients can be seen in PAH.
V/Q Scans are done to rule out chronic thromboembolic pulmonary hypertension (CTEPH) once the common causes are excluded. Patients with perfusion defects are further evaluated by CT pulmonary angiography and right heart catheterization.[24][25][26][27]
Pulmonary hypertension diagnosis can be confirmed with right heart catheterization that measures the hemodynamic parameters accurately. Pulmonary artery catheter can directly measure central venous pressure, right heart intracardiac pressure, pulmonary artery pressure, pulmonary capillary wedge pressure/PA occlusion pressure, cardiac output, mixed venous oxyhemoglobin saturation. These direct measures such as mean PAP, pulmonary capillary wedge pressure (PCWP) and, cardiac output (CO) can be used to calculate pulmonary vascular resistance indirectly. Pulmonary vascular resistance (PVR) helps in determining the prognosis of pulmonary hypertension. Mean PAP is high in both PAH and Group 2 PH, but PCWP greater than 15 mm Hg is typically seen in PH due to left heart disease.[28][29][30][31]
Treatment / Management
Before initiating the therapy, baseline disease severity should be assessed. Disease severity is determined by assessing functional impairment based on exercise capacity and hemodynamic derangement.
The goal of the management of pulmonary hypertension is primary therapy to treat the underlying cause of pulmonary hypertension. Following primary therapy, the disease severity is reassessed.
Patients with WHO Functional Class II, III and, IV with persistent pulmonary hypertension despite primary therapy require advanced therapy which targets pulmonary hypertension itself and not the underlying cause). Advanced therapy is used in most Group 1 PAH cases and some patients with group 3, 4, and 5 on a case-by-case basis. However, it is almost never used for Group 2 patients.
Advanced therapy includes prostacyclin agonists, endothelin receptor antagonists, NO-cGMP enhancers, and rarely, calcium channel blockers.
A combination or alternate therapy is used in patients with refractory PAH.
Group 1 PH: None of the primary therapies have been proven to be successful for most types. Often, advanced therapy is required.
Group 2 PH: Medications or valve repair to optimize left heart function in these patients. In patients with volume overload, diuretics can be considered. Thiazides followed by loop diuretics can be used. However, over diuresis should be avoided. Sodium restriction can be helpful.
Heart failure should be managed according to ejection fraction. Beta-blockers, nitrates, and digoxin should be avoided in a preserved ejection fraction, until very necessary. Treatment of coexisting conditions (hypertension, valvular heart disease, ischemic heart disease, diabetes, thyroid disease) and lifestyle modification. Remote monitoring strategies such as weight and symptoms can help reduce hospitalizations in heart failure patients with reduced or preserved ejection fraction.
Group 3 PH: Supplemental oxygen to correct hypoxemia and management of the underlying cause of hypoxemia.
Group 4 PH: The most effective step in effectively reducing the resistance of pulmonary vessels secondary to chronic thrombo-emboli is pulmonary thromboendarterectomy. Small vessel disease is a contraindication as the reduction in the pulmonary vascular resistance post-thromboendarterectomy is not as expected. Pulmonary artery steal syndrome and, reperfusion pulmonary edema are the common negative sequelae. After initial improvement post-thromboendarterectomy, late adverse events such as residual pulmonary hypertension and recurrent pulmonary hypertension can take place.
If the disease is inoperable and thrombectomy cannot be performed, Balloon pulmonary-artery angioplasty is an alternative.
If the thromboembolism is surgically inaccessible or there is recurrent or persistent pulmonary hypertension post-thromboendarterectomy, advanced medical therapy can be considered. Advanced medical therapy includes anticoagulants and pulmonary vasodilators. However, there is no approved medical therapy.[32][33][34][35]
Heart-lung transplantation can be considered and is the final treatment for those patients who fail to respond to the above measures.[36][37][38]
Differential Diagnosis
The non-specific clinical picture leads to a delay in the diagnosis of pulmonary hypertension due to overlap with common heart and lung diseases. The diagnosis of pulmonary hypertension can be made by evaluating and excluding the following-
- Anemia
- Chronic obstructive pulmonary disease (COPD)
- Pulmonary fibrosis
- Chronic obstructive asthma
- Obstructive sleep apnea
- Cor pulmonale
- Dilated Cardiomyopathy
- Heart failure
- Coronary artery disease
- Mitral stenosis
- Connective tissue diseases
- Portal hypertension
- Liver disease
- Budd-Chiari syndrome
- Pulmonary embolism
- Hypothyroidism
Prognosis
The mean survival of people with evidence of right heart failure or severe pulmonary hypertension (greater than 55 mm Hg mean pulmonary artery pressure) is about a year. People with preserved right heart function and with mean pulmonary artery pressure less than 55 mm Hg survive for approximately 3 years.
Untreated IPAH patients are known to have a median survival of 2 to 3 years.[39][21]
Complications
Complications of pulmonary hypertension include:[40]
- Cor pulmonale and right heart failure
- Alveolar hemorrhage
- Pericardial effusion and tamponade
- Pulmonary cavitation
Deterrence and Patient Education
Patient education on compliance with therapies for secondary causes of pulmonary hypertension is of utmost importance in preventing this disease. patients diagnosed with obstructive sleep apnea, chronic obstructive pulmonary disease, and venous thromboembolism need to be counselled on the potential risk of pulmonary hypertension if the underlying condition is not treated properly. Smoking cessation should be especially emphasized in all patients with diagnosed pulmonary hypertension.
Pearls and Other Issues
- Chronic lung diseases are a common cause of pulmonary hypertension but it almost never increases the pulmonary artery pressure more than 50 mm Hg and in most cases only causes mild pulmonary hypertension. Therefore, evaluate for other causes if pressure is markedly increased and goes beyond 50 mm Hg.
- Evaluate for collagen vascular diseases as interstitial lung disease (ILD) coexists with many of them such as CREST syndrome, scleroderma, systemic lupus erythematosus (SLE), Sjogren syndrome, dermatomyositis, polymyositis, and rheumatoid arthritis.
- Some patients with obstructive sleep apnea may have mild pulmonary hypertension. Alveolar hypoventilation may occur due to deformities in the thoracic vertebra, neuromuscular diseases such as paralysis of the diaphragm or weak respiratory muscles, trauma to the phrenic nerve, all of these can lead to pulmonary hypertension. Supplemental oxygen and intermittent PPV is helpful.
- Thyroid disease is a risk factor for both chronic thromboembolism and idiopathic.
- Progressive dyspnea and clinical features of pulmonary hypertension in sarcoidosis patients is an indication for a thorough evaluation. Sarcoidosis causes severe chronic fibrocystic changes in the lung and can affect the cardiovascular system directly. These changes can lead to pulmonary hypertension. Severe pulmonary hypertension in sarcoidosis is due to the direct involvement of pulmonary vessels. Intravenous epoprostenol has shown a favorable response in many such patients.
- Schistosomiasis is rare in the United States of America, but it is the most common cause of pulmonary hypertension worldwide. The ova in the lungs can be due to the embolization from the liver which causes the inflammatory changes in pulmonary vasculature that leads to chronic changes. Pulmonary hypertension in schistosomiasis occurs in addition to portal hypertension and hepatosplenic disease. Parasite ova in the stools or urine of patients with symptoms confirms the diagnosis of pulmonary hypertension due to schistosomiasis. The therapy for pulmonary hypertension due to schistosomiasis is unknown.[3][41][42][43]
Enhancing Healthcare Team Outcomes
Secondary pulmonary hypertension is a complex, life-threatening disease that significantly affects the quality of life and over time leads to right heart failure. A timely evaluation, proper treatment, regular follow up and patient education can positively affect the outcome of the disease. For this, integrated, interprofessional care with the involvement of different subspecialties is required. The physicians with a team of nurse specialists, pharmacists and, social workers coordinate closely to provide the best possible care and counsel these patients.
Non-specific symptoms of the disease usually result in a delay in diagnosis. The family physician suspects the disease based on non-invasive tests (echocardiogram, x-ray). Once the presumptive diagnosis of pulmonary hypertension has been made, it is very critical to classify the type of pulmonary hypertension as the treatment for one group may cause deleterious effects if used for another group. These patients are, therefore, referred to different subspecialty physicians to determine the cause. A referral to the pulmonologist is essential for sleep studies and pulmonary function tests to exclude lung disease as a cause. To rule out left heart disease, patients are referred to a cardiologist. Right heart catheterization to confirm the diagnosis is also done by the cardiologist. Similarly, to rule out other causes the patients are also referred to rheumatologists, and, other doctors. It is also important to counsel the patients so that they can live with this disease.
Treatment of the cause of pulmonary hypertension is the key to manage the disease. Hence, collaboration with physicians, physician assistants, and nurse practitioners in different specialties is essential to manage secondary pulmonary hypertension. [Level V]
Media
(Click Image to Enlarge)
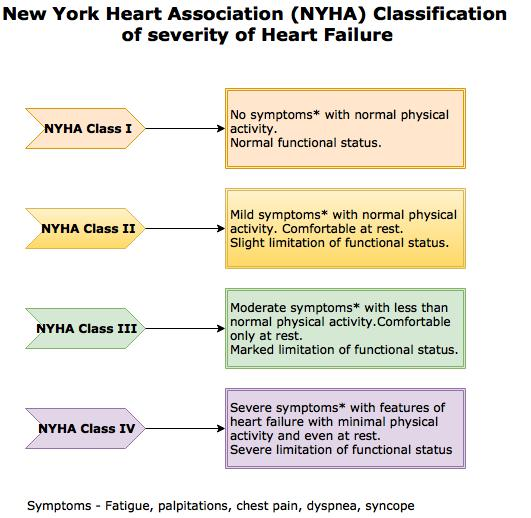
NYHA Classification - Heart failure Contributed by the New York Health Association (NYHA)
References
Ayach B, Fine NM, Rudski LG. Right ventricular strain: measurement and clinical application. Current opinion in cardiology. 2018 Sep:33(5):486-492. doi: 10.1097/HCO.0000000000000540. Epub [PubMed PMID: 30063529]
Level 3 (low-level) evidenceBrinkman JE, Sharma S. Physiology, Pulmonary. StatPearls. 2023 Jan:(): [PubMed PMID: 29494033]
Charles L, Triscott J, Dobbs B. Secondary Hypertension: Discovering the Underlying Cause. American family physician. 2017 Oct 1:96(7):453-461 [PubMed PMID: 29094913]
Maeder MT, Weber L, Buser M, Gerhard M, Haager PK, Maisano F, Rickli H. Pulmonary Hypertension in Aortic and Mitral Valve Disease. Frontiers in cardiovascular medicine. 2018:5():40. doi: 10.3389/fcvm.2018.00040. Epub 2018 May 23 [PubMed PMID: 29876357]
McGee M, Whitehead N, Martin J, Collins N. Drug-associated pulmonary arterial hypertension. Clinical toxicology (Philadelphia, Pa.). 2018 Sep:56(9):801-809. doi: 10.1080/15563650.2018.1447119. Epub 2018 Mar 6 [PubMed PMID: 29508628]
Rezaee ME, Nichols EL, Sidhu M, Brown JR. Combined Post- and Precapillary Pulmonary Hypertension in Patients With Heart Failure. Clinical cardiology. 2016 Nov:39(11):658-664. doi: 10.1002/clc.22579. Epub 2016 Oct 21 [PubMed PMID: 27768231]
Ponomarev RV, Model SV, Averbukh OM, Gavrilov AM, Galstyan GM, Lukina EA. [Progressive pulmonary hypertension in a patient with type 1 Gaucher disease]. Terapevticheskii arkhiv. 2017:89(10):71-74. doi: 10.17116/terarkh2017891071-74. Epub [PubMed PMID: 29171474]
Kovacs G, Avian A, Wutte N, Hafner F, Moazedi-Fürst F, Kielhauser S, Aberer E, Brodmann M, Graninger W, Foris V, Olschewski A, Olschewski H. Changes in pulmonary exercise haemodynamics in scleroderma: a 4-year prospective study. The European respiratory journal. 2017 Jul:50(1):. pii: 1601708. doi: 10.1183/13993003.01708-2016. Epub 2017 Jul 13 [PubMed PMID: 28705939]
Brida M, Gatzoulis MA. Pulmonary arterial hypertension in adult congenital heart disease. Heart (British Cardiac Society). 2018 Oct:104(19):1568-1574. doi: 10.1136/heartjnl-2017-312106. Epub 2018 May 2 [PubMed PMID: 29720395]
Ribeiro Baptista B, Petitpain N, Gomez E, Yelehé-Okouma M, Valentin S, Guillaumot A, Chabot F, Chaouat A. Pulmonary arterial hypertension in patient treated for multiple sclerosis with 4-aminopyridine. Fundamental & clinical pharmacology. 2019 Feb:33(1):127-129. doi: 10.1111/fcp.12396. Epub 2018 Jul 15 [PubMed PMID: 29956855]
Kolosionek E, King J, Rollinson D, Schermuly RT, Grimminger F, Graham BB, Morrell N, Butrous G. Schistosomiasis causes remodeling of pulmonary vessels in the lung in a heterogeneous localized manner: Detailed study. Pulmonary circulation. 2013 Apr:3(2):356-62. doi: 10.4103/2045-8932.114764. Epub [PubMed PMID: 24015336]
Goyal VK, Solanki SL, Baj B. Pulmonary hypertension and post-operative outcome in renal transplant: A retrospective analysis of 170 patients. Indian journal of anaesthesia. 2018 Feb:62(2):131-135. doi: 10.4103/ija.IJA_529_17. Epub [PubMed PMID: 29491519]
Level 2 (mid-level) evidenceFuloria M, Aschner JL. Persistent pulmonary hypertension of the newborn. Seminars in fetal & neonatal medicine. 2017 Aug:22(4):220-226. doi: 10.1016/j.siny.2017.03.004. Epub 2017 Mar 23 [PubMed PMID: 28342684]
Zhang Q, Wang L, Zeng H, Lv Y, Huang Y. Epidemiology and risk factors in CKD patients with pulmonary hypertension: a retrospective study. BMC nephrology. 2018 Mar 20:19(1):70. doi: 10.1186/s12882-018-0866-9. Epub 2018 Mar 20 [PubMed PMID: 29554879]
Level 2 (mid-level) evidenceSikachi RR, Sahni S, Mehta D, Agarwal A, Agrawal A. Nationwide trends in inpatient admissions of pulmonary hypertension in the United States from 2000 to 2013. Advances in respiratory medicine. 2017:85(2):77-86. doi: 10.5603/ARM.2017.0014. Epub [PubMed PMID: 28440533]
Level 3 (low-level) evidencede-Miguel-Díez J, López-de-Andrés A, Hernandez-Barrera V, Jimenez-Trujillo I, de-Miguel-Yanes JM, Mendez-Bailón M, Jimenez-Garcia R. National trends and outcomes of hospitalizations for pulmonary hypertension in Spain (2001-2014). International journal of cardiology. 2018 Jul 15:263():125-131. doi: 10.1016/j.ijcard.2018.04.026. Epub 2018 Apr 7 [PubMed PMID: 29673852]
Naeije R, Vanderpool R, Peacock A, Badagliacca R. The Right Heart-Pulmonary Circulation Unit: Physiopathology. Heart failure clinics. 2018 Jul:14(3):237-245. doi: 10.1016/j.hfc.2018.02.001. Epub [PubMed PMID: 29966623]
Naeije R, Gerges M, Vachiery JL, Caravita S, Gerges C, Lang IM. Hemodynamic Phenotyping of Pulmonary Hypertension in Left Heart Failure. Circulation. Heart failure. 2017 Sep:10(9):. pii: e004082. doi: 10.1161/CIRCHEARTFAILURE.117.004082. Epub [PubMed PMID: 28912263]
Yang W, Marsden AL, Ogawa MT, Sakarovitch C, Hall KK, Rabinovitch M, Feinstein JA. Right ventricular stroke work correlates with outcomes in pediatric pulmonary arterial hypertension. Pulmonary circulation. 2018 Jul-Sep:8(3):2045894018780534. doi: 10.1177/2045894018780534. Epub 2018 May 16 [PubMed PMID: 29767574]
Yandrapalli S, Tariq S, Kumar J, Aronow WS, Malekan R, Frishman WH, Lanier GM. Chronic Thromboembolic Pulmonary Hypertension: Epidemiology, Diagnosis, and Management. Cardiology in review. 2018 Mar/Apr:26(2):62-72. doi: 10.1097/CRD.0000000000000164. Epub [PubMed PMID: 28832374]
Vanderpool RR, Saul M, Nouraie M, Gladwin MT, Simon MA. Association Between Hemodynamic Markers of Pulmonary Hypertension and Outcomes in Heart Failure With Preserved Ejection Fraction. JAMA cardiology. 2018 Apr 1:3(4):298-306. doi: 10.1001/jamacardio.2018.0128. Epub [PubMed PMID: 29541759]
Florentin J, Coppin E, Vasamsetti SB, Zhao J, Tai YY, Tang Y, Zhang Y, Watson A, Sembrat J, Rojas M, Vargas SO, Chan SY, Dutta P. Inflammatory Macrophage Expansion in Pulmonary Hypertension Depends upon Mobilization of Blood-Borne Monocytes. Journal of immunology (Baltimore, Md. : 1950). 2018 May 15:200(10):3612-3625. doi: 10.4049/jimmunol.1701287. Epub 2018 Apr 9 [PubMed PMID: 29632145]
Zhang M, Feng Z, Huang R, Sun C, Xu Z. Characteristics of Pulmonary Vascular Remodeling in a Novel Model of Shunt-Associated Pulmonary Arterial Hypertension. Medical science monitor : international medical journal of experimental and clinical research. 2018 Mar 19:24():1624-1632 [PubMed PMID: 29554080]
Calcutteea A, Chung R, Lindqvist P, Hodson M, Henein MY. Differential right ventricular regional function and the effect of pulmonary hypertension: three-dimensional echo study. Heart (British Cardiac Society). 2011 Jun:97(12):1004-11. doi: 10.1136/hrt.2010.208900. Epub 2011 Apr 12 [PubMed PMID: 21487122]
Assad TR, Maron BA, Robbins IM, Xu M, Huang S, Harrell FE, Farber-Eger EH, Wells QS, Choudhary G, Hemnes AR, Brittain EL. Prognostic Effect and Longitudinal Hemodynamic Assessment of Borderline Pulmonary Hypertension. JAMA cardiology. 2017 Dec 1:2(12):1361-1368. doi: 10.1001/jamacardio.2017.3882. Epub [PubMed PMID: 29071338]
Goerne H, Batra K, Rajiah P. Imaging of pulmonary hypertension: an update. Cardiovascular diagnosis and therapy. 2018 Jun:8(3):279-296. doi: 10.21037/cdt.2018.01.10. Epub [PubMed PMID: 30057876]
Chaouat A, Cherifi A, Sitbon O, Girerd N, Zysman M, Faure M, Mandry D, Mercy M, Guillaumot A, Fay R, Marie PY, Chabot F. [Evaluation of cardiac MRI in the follow up assessment of patients with pulmonary arterial hypertension]. Revue des maladies respiratoires. 2018 Sep:35(7):749-758. doi: 10.1016/j.rmr.2018.01.010. Epub 2018 Jun 23 [PubMed PMID: 29945811]
Grosse A, Grosse C, Lang IM. Distinguishing Chronic Thromboembolic Pulmonary Hypertension From Other Causes of Pulmonary Hypertension Using CT. AJR. American journal of roentgenology. 2017 Dec:209(6):1228-1238. doi: 10.2214/AJR.17.17871. Epub 2017 Oct 5 [PubMed PMID: 28981358]
Wilkens H, Held M. [Heart or lung? : Diagnostics and management of unclear exertional dyspnea]. Herz. 2018 Sep:43(6):567-582. doi: 10.1007/s00059-018-4730-2. Epub [PubMed PMID: 30027500]
Okumus G, Aslan GK, Arseven O, Ongen G, Issever H, Kiyan E. The role of an activity monitor in the objective evaluation of patients with pulmonary hypertension. The clinical respiratory journal. 2018 Jan:12(1):119-125. doi: 10.1111/crj.12495. Epub 2016 May 23 [PubMed PMID: 27149246]
Rosenkranz S, Preston IR. Right heart catheterisation: best practice and pitfalls in pulmonary hypertension. European respiratory review : an official journal of the European Respiratory Society. 2015 Dec:24(138):642-52. doi: 10.1183/16000617.0062-2015. Epub [PubMed PMID: 26621978]
Menon K, Sutphin PD, Bartolome S, Kalva SP, Ogo T. Chronic thromboembolic pulmonary hypertension: emerging endovascular therapy. Cardiovascular diagnosis and therapy. 2018 Jun:8(3):272-278. doi: 10.21037/cdt.2018.06.07. Epub [PubMed PMID: 30057875]
Segura-Ibarra V, Wu S, Hassan N, Moran-Guerrero JA, Ferrari M, Guha A, Karmouty-Quintana H, Blanco E. Nanotherapeutics for Treatment of Pulmonary Arterial Hypertension. Frontiers in physiology. 2018:9():890. doi: 10.3389/fphys.2018.00890. Epub 2018 Jul 13 [PubMed PMID: 30061840]
Ahmad KA, Banales J, Henderson CL, Ramos SE, Brandt KM, Powers GC. Intravenous epoprostenol improves oxygenation index in patients with persistent pulmonary hypertension of the newborn refractory to nitric oxide. Journal of perinatology : official journal of the California Perinatal Association. 2018 Sep:38(9):1212-1219. doi: 10.1038/s41372-018-0179-7. Epub 2018 Jul 25 [PubMed PMID: 30046179]
Pedersen J, Hedegaard ER, Simonsen U, Krüger M, Infanger M, Grimm D. Current and Future Treatments for Persistent Pulmonary Hypertension in the Newborn. Basic & clinical pharmacology & toxicology. 2018 Oct:123(4):392-406. doi: 10.1111/bcpt.13051. Epub 2018 Jul 19 [PubMed PMID: 29855164]
Alfraidi H, Qanash S, Bshouty Z. Pulmonary Arterial Hypertension Specific Therapy in Patients with Combined Post- and Precapillary Pulmonary Hypertension. Pulmonary medicine. 2018:2018():7056360. doi: 10.1155/2018/7056360. Epub 2018 Mar 1 [PubMed PMID: 29686899]
Westerhof BE, Saouti N, van der Laarse WJ, Westerhof N, Vonk Noordegraaf A. Treatment strategies for the right heart in pulmonary hypertension. Cardiovascular research. 2017 Oct 1:113(12):1465-1473. doi: 10.1093/cvr/cvx148. Epub [PubMed PMID: 28957540]
Pahal P, Sharma S. Idiopathic Pulmonary Artery Hypertension. StatPearls. 2023 Jan:(): [PubMed PMID: 29489262]
Calcaianu G, Calcaianu M, Gschwend A, Canuet M, Meziani F, Kessler R. Hemodynamic profile of pulmonary hypertension (PH) in ARDS. Pulmonary circulation. 2018 Jan-Mar:8(1):2045893217753415. doi: 10.1177/2045893217753415. Epub 2017 Dec 28 [PubMed PMID: 29283029]
Mak SM, Strickland N, Gopalan D. Complications of pulmonary hypertension: a pictorial review. The British journal of radiology. 2017 Feb:90(1070):20160745. doi: 10.1259/bjr.20160745. Epub 2016 Dec 7 [PubMed PMID: 27925469]
Rashidi F, Sate H, Faraji E, Tahsini Tekantapeh S. Thyrotoxicosis presenting as exertional dyspnea and pulmonary hypertension: Case report and review of literature. SAGE open medical case reports. 2017:5():2050313X17715584. doi: 10.1177/2050313X17715584. Epub 2017 Jun 19 [PubMed PMID: 28680634]
Level 3 (low-level) evidenceShujaat A, Bellardini J, Cury JD, Bajwa AA. Use of pulmonary arterial hypertension-specific therapy in non-WHO group I pulmonary hypertension. Southern medical journal. 2015 Jan:108(1):51-7. doi: 10.14423/SMJ.0000000000000217. Epub [PubMed PMID: 25580759]
Level 2 (mid-level) evidenceKolosionek E, Crosby A, Harhay MO, Morrell N, Butrous G. Pulmonary vascular disease associated with schistosomiasis. Expert review of anti-infective therapy. 2010 Dec:8(12):1467-73. doi: 10.1586/eri.10.124. Epub [PubMed PMID: 21133670]
Level 3 (low-level) evidence