Introduction
Bone is a unique connective tissue type, being one that undergoes mineralization. Bone's inorganic component consists of calcium hydroxyapatite, which imparts strength and toughness. This mineral is also the body's main calcium (99%) and phosphate (85%) reservoir and stores 65% of its sodium and magnesium.
Meanwhile, bone's organic component is comprised of bone cells and matrix proteins. The bone cells include the following:
- Osteoprogenitor cells: pluripotent mesenchymal stem cells in bone surfaces that growth factors can stimulate to differentiate into osteoblasts
- Osteoblasts: initiate mineralization and produce, transport, and arrange matrix proteins. Osteoblast actions are regulated by parathyroid hormone, vitamin D, estrogen, growth factors, cytokines, leptin, and low-density lipoprotein-related protein 5. Osteoblasts may become osteocytes or surface-lining cells of bones.
- Osteocytes: the most numerous cells in bone. Osteocytes participate in key bodily functions like calcium and phosphate homeostasis and cyclic adenosine monophosphate activation.
- Osteoclast: hematopoietic progenitor cell-derived bone cells responsible for bone resorption
Bone matrix proteins include type 1 collagen and noncollagenous osteoblast-derived proteins.
Homeobox genes encode the skeletal morphogenesis regulators. Mesenchyme from the following regions gives rise to different parts of the skeleton:
- Cranial neural crest: craniofacial skeleton
- Paraxial mesoderm: axial skeleton
- Lateral plate mesoderm: appendicular skeleton
In the embryo, craniofacial and clavicular bones undergo intramembranous ossification, whereby osteoblasts form bone directly from mesenchyme. These bones enlarge only when new bone deposits on the surface—a process called "appositional growth." Meanwhile, most other bones begin as a cartilage anlage, undergoing enchondral ossification around the 8th week of gestation. Bones that form this way increase in length due to the development of the physis or growth plate at their ends. Growth plates have a cartilaginous core covered by a layer of bone (primary spongiosa). Enchondral ossification within growth plates increases bone length and diameter.
Hyaline-cartilage bone tumors arise either within the medullary cavity or on the bone surface. Enchondromas are benign medullary cavity hyaline-cartilage tumors occurring in bones of endochondral origin.[1] These growths are usually solitary, central, metaphyseal tubular bone lesions. In contrast, subperiosteal or juxtacortical chondromas form on the bone surface.
Enchondromas arise most commonly from the hand and foot bones but may occasionally appear in the femur and humerus. These neoplasms are also the most common primary bone tumors of the hand, where they commonly appear in the proximal phalanges. Other common sites include the middle phalanges, metacarpals, and distal phalanges.[2][3]
Enchondromatous tumors typically begin and grow in childhood, arising from rests of growth plate cartilage or chondrocytes that initially proliferate but stop developing normally and persist throughout adulthood.[4] These tumors are most frequently noted in the 3rd and 4th decades of life. The small finger is the most commonly affected digit.[5] Hand enchondromas are unique, as they may also demonstrate cellular atypia, resembling chondrosarcoma on histopathological examination.[6]
Enchondromas have malignant potential and may transform into chondrosarcoma.[7] However, malignancies arising from enchondromas are low-grade and rarely metastasize to other body regions.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The persistence of growth plate chondrocytes in bone leads to enchondroma formation.[8] However, the reason for chondrocyte persistence is unknown. Genetic mutations are rare and often sporadic if involved.
Somatic isocitrate dehydrogenase-1 (IDH1) and 2 (IDH2) gene mutations have been implicated. Such mutations lead to the synthesis of defective isocitrate dehydrogenase—a tricarboxylic acid cycle enzyme that catalyzes isocitrate conversion to α-ketoglutarate.[9][10] The mutated enzyme catalyzes α-ketoglutarate reduction to the oncometabolite D-2-hydroxyglutarate (D-2-HG). D-2-HG competitively inhibits α-ketoglutarate-dependent enzymes, leading to DNA hypermethylation and histone modification.[11] These processes promote cartilaginous tumor formation and prevent normal osteogenic differentiation of mesenchymal stem cells.[12]
Somatic parathyroid hormone 1 receptor (PTHR1) gene mutations have also been found in enchondromas. The parathyroid hormone 1 receptor signaling pathway is a key regulator of enchondral ossification. Additionally, the Indian Hedgehog pathway is constitutively active in some cases of enchondromas. Wnt/β-catenin signaling downregulates Indian Hedgehog activity in enchondromas. Thus, the Wnt/β-catenin signaling pathway is a potential therapeutic target in treating enchondromas.[13]
Epidemiology
Enchondromas are the most prevalent intraosseous cartilage neoplasms, accounting for approximately 3% of bone tumors and 13% of benign bone growths. Most appear in the small hand and foot bones, but femoral and humeral enchondromas have also been documented.[14]
Enchondromatosis is the growth of multiple enchondromas. Ollier disease is a condition arising from enchondromatosis with an asymmetric distribution in the appendicular skeleton. This bone disorder has an estimated prevalence of 1 in 100,000.[15]
Maffucci syndrome is a condition presenting with enchondromatosis and multiple soft tissue hemangiomas and lymphangiomas. This disorder is rare, with fewer than 200 cases reported in the literature.[16] Although histologically similar to a solitary enchondroma, the cytological changes in this syndrome appear more bizarre.
Both Ollier disease and Maffucci syndrome are associated with an increased risk of chondrosarcoma and visceral malignancies.
Pathophysiology
Terminal chondrocyte differentiation in the growth plate is dysregulated in enchondroma formation. Most enchondromas begin in the medullary portion of the diaphysis. The tumors arise from ectopic cartilaginous nests in the metaphyseal region and expand outward toward the cortex. Enlarging lesions may cause a pathologic fracture.[17]
The enchondromatous tumors of Ollier disease are typically located at the epiphysis, metaphysis, and diaphysis, potentially hindering normal bone growth. Limb shortening may occur due to epiphyseal invasion, metaphyseal widening, and long-bone bowing.[18]
Histopathology
On gross examination, the typical enchondroma is smaller than 3 centimeters. Microscopically, enchondromas appear as gray-blue, translucent, hypocellular, avascular tumors with abundant hyaline cartilage. The nuclei of these cells are fairly regular, with few mitotic figures. Juxtacortical chondromas and enchondromas in regions like the hands may be hypercellular and atypical but remain benign.[19]
Chondroid matrix punctate calcifications define the microscopic appearance of enchondromas. The tumor is composed of well-circumscribed, benign hyaline cartilage nodules. Limited engulfment of the adjacent lamellar and cortical bone is seen. Chondrocyte nuclei are small and uniformly round with condensed chromatin. Binucleate forms are rare. Endochondral ossification foci may be found in heavily calcified enchondromas.
Long bone enchondromas often appear benign microscopically but can recur after removal. As with many tumors, staging systems are used to classify the tumor further. Bone sarcoma staging follows the tumor, node, and metastasis (TNM) guidelines.[20]
The enchondromas in syndromes like Ollier disease and Maffucci syndrome exhibit more cellularity and atypia than the classic, single enchondroma.[21] The increased cellularity and atypia make distinguishing enchondromatosis tumors from chondrosarcoma more difficult, especially when the malignancy is in its early stages. Benign, atypical cartilaginous lesions and chondrosarcoma grade 1 are nearly indistinguishable. However, cartilaginous lesions are typically hypocellular, while chondrosarcoma grade 1 has a hypercellular appearance. Additionally, chondrosarcoma exhibits more binucleated chondrocytes, pleomorphism, cortical destruction, host bone engulfment, and irregular cellular distribution.
History and Physical
Enchondromas may manifest at any age. However, the onset of presentation of the solitary enchondroma is classically in the 2nd or 3rd decade of life. Enchondromatosis syndromes become symptomatic earlier, typically before age 10.[22] The tumor is often discovered after the patient develops a pathologic fracture. Pain, swelling, and deformity are common. Pathologic fractures are seen in 40-60% of patients at presentation.[23] Others are detected incidentally on radiographic imaging.[24]
The classic location of the lesions is in the proximal metaphyses of proximal phalanges, with a predilection for the ulnar side of the bones.[25] Distal phalangeal enchondromas may have associated nail deformities.
A thorough examination of the hand and wrist is necessary for patients with hand enchondromas. Passive movements of the interphalangeal and metacarpophalangeal joints need to be evaluated. Flexor and extensor tendon function must also be assessed, as distal phalangeal tumors can cause flexor tendon avulsion.
The skin, chest, abdomen, and neuromuscular function must be fully evaluated in patients with suspected enchondromatosis syndromes. These individuals are at risk of developing multiple hemangiomas and ovarian and brain malignancies.
Evaluation
Plain hand radiographs with posteroanterior, lateral, and oblique views should be obtained during the initial evaluation of hand enchondromas (see Image. Enchondroma Radiography). These tumors have varied radiographic appearances, influenced by location and extent of calcification. Enchondromas typically appear as well-defined, solitary metaphyseal defects, especially in the long bones. Centrally located lesions usually appear as well-circumscribed areas of rarefaction, most frequently diaphyseal, with an expanded surrounding cortex. Juxtacortical lesions are eccentric. located beneath the periosteum in well-defined cortical defects. Small, flocculent foci of calcification are visible within the tumors. Some may resemble medullary bone infarcts.[26]
Radiographically visible calcifications usually appear as fine, punctate stipplings. A bone infarct may be present if these areas are more pronounced. Some calcifications appear as rings. Larger lesions can cause endosteal scalloping and cortical expansion and thinning. Heavily calcified lesions may resemble bone infarction or bone islands, while an unclassified lesion may appear lytic. Cortical thinning or breach, tumor expansion, and soft tissue involvement are associated with advanced lesions or chondrosarcoma.[27]
Computed tomography (CT) is useful in evaluating matrix mineralization and cortex integrity. Magnetic resonance imaging (MRI) helps identify aggressive and destructive features. Enchondroma and chondrosarcoma often appear similarly on first-pass analysis. Both exhibit low signal intensities on T1 and reciprocal high-intensity changes with a lobular growth pattern on T2-weighted imaging. Both show enhancement with gadolinium contrast in peripheral and spatial areas.
Neither dynamic contrast-enhanced MRI nor advanced techniques like diffusion-weighted imaging and hydrogen proton spectroscopy have proven effective in differentiating enchondroma from chondrosarcoma. However, peritumoral edema may differentiate these tumors. A case series analysis revealed that chondrosarcoma is more likely to have peritumoral edema than enchondroma. Both CT and MRI can easily distinguish bone infarct from enchondroma.
Evidence of a lesion in a radiologic study is often insufficient to diagnose an enchondroma. Diagnostic confirmation requires histopathologic examination of the biopsied lesion. However, differentiating between benign and malignant hyaline cartilage lesions poses an even greater challenge. In some cases, the diagnosis may remain in question despite extensive tissue examination unless the suspicion of enchondroma or early chondrosarcoma is strong. Indicators of potential malignancy include the following:[28]
- Large size
- Presence of a large unmineralized component
- Significant thinning of the adjacent cortex
- Bone scan activity greater than that of the anterior superior iliac spine
- Progressive chondrite matrix destruction due to an expanding non-mineralized component
- Rapidly enlarging, painful lesion
- Expansile soft tissue mass
Only 2 known biomarkers can distinguish enchondroma from chondrosarcoma: periostin and α-methylacyl-CoA racemase (AMACR). Periostin, a stroma-related protein, is reportedly absent in enchondroma but present in low-grade chondrosarcoma.[29] Meanwhile, AMACR, a mitochondrial and peroxisomal enzyme, is often expressed by enchondroma but rarely by chondrosarcoma.[30] Further investigation is needed to validate the reliability of these biomarkers in enchondroma and chondrosarcoma diagnosis.
Treatment / Management
Currently, no standardized algorithm for enchondroma surgery exists. Asymptomatic enchondromas may be observed, though routine radiographic follow-up for asymptomatic lesions is usually unnecessary. Symptomatic lesions and tumors producing pathologic fractures often improve with surgery. Operative management also helps establish a definitive diagnosis. However, the optimal timing of surgical intervention is unknown. Early and delayed surgical intervention was shown to have similar functional outcomes.
The surgical technique typically used for treating symptomatic enchondroma is simple curettage with bone grafting. The bone graft may be allogeneic, autogenous, or synthetic. The graft type's impact on healing, recurrence, complications, and malignant transformation is unknown. The necessity of curettage with grafting remains unproven.
Care must be taken to prevent tumor dissemination during surgery. Tourniquet control without exsanguination using an esmarch bandage is performed during the procedure. Adjuncts used to kill the residual tumor cells and help reduce recurrence include phenol, liquid nitrogen, cryotherapy, polymethyl methacrylate, and electrocautery.[31][32] However, no data suggests which adjunct is superior among the various treatment options. A frozen section is not recommended in these procedures.(B2)
The bone void can be filled using an autograft, allograft, bone substitute, or bone cement after curettage. However, some studies have demonstrated that leaving the void unfilled renders equivalent functional outcomes. This technique saves surgical time, prevents donor site morbidity, and minimizes foreign body-induced complications, as when allografts and bone cement are used.
Fracture prophylaxis by internal fixation methods is controversial. Previous guidelines based on retrospective studies were limited by the use of plain radiographs, subjective patient information, and inadequate understanding of the biomechanical factors involved in the neoplastic process. The Mirels criteria were developed to quantify bone neoplasm-related fracture risk. These criteria consider the location, pain, and lesion type and size.[33] Scores greater than 8 indicate a significant fracture risk. Thus, prophylactic internal fixation may be recommended in such cases. Lesions with scores less than 7 have a reduced fracture risk and may simply be observed.
A 23-year retrospective review of enchondroma patients reported that enchondroma treatment with biopsy, curettage, and allograft implantation resulted in a full range of motion in more than 80% of the patients. Additionally, recurrence was rarely seen and was more common with Ollier's disease and giant-form lesions. Preoperative pathologic fractures are associated with a greater risk of postoperative extension deficit.
Various surgical techniques have been used for enchondroma treatment. One case report described a successful endoscopic curettage and bone grafting of a proximal finger phalangeal enchondroma.[34] Another case series reported bone fixation with headless intramedullary screws after long-hand bone enchondroma resection with good results.[35] A 10-year retrospective cohort study found that osteoscopic surgery leads to earlier functional recovery and fewer complications than open surgery.[36] Radiofrequency ablation was recently shown to have value in treating flat pelvic bone enchondromas.[37](B2)
Differential Diagnosis
Conditions that may present similarly to enchondromatous lesions include the following:[38][39]
- Bone infarction, which has a similar radiographic appearance
- Tuberculous dactylitis, due to bone pain in the absence of a fracture
- Low-grade chondrosarcoma, also due to non-fracture-related bone pain
- Other benign neoplasms like giant cell reparative granuloma, giant cell tumors, and chondroblastoma
These conditions may be differentiated from enchondromas with a good clinical evaluation, appropriate laboratory and imaging studies, and histopathology.
Staging
Takigawa proposed a radiographic classification for enchondromas, which had 5 morphological categories:
- Type A: central
- Type B: eccentric
- Type C: associated
- Type D: polycentric
- Type E: giant form
One study found that the Takigawa classification could predict the optimal enchondroma treatment option.[44] However, more large-scale studies are necessary to evaluate the reliability of this system in predicting prognosis and surgical outcomes of enchondromatous growths.
Prognosis
Solitary enchondromatous lesions are typically self-limited. Recurrence is rare following curettage and bone grafting. However, giant-form tumors and long bone involvement are associated with a higher recurrence risk. Meanwhile, enchondromatosis increases not only the risk of recurrence but also the probability of malignant transformation.
Complications
The potential complications of delayed enchondroma treatment include the following:
- Intramedullary enchondroma growth and cortical bone encroachment may produce a pathologic fracture.
- Enchondromas may interfere with proper bone development in children.
- Pathologic fractures requiring primary surgery increase hospitalization costs.
- Delayed surgery of pathological fractures, ie, when surgical intervention is postponed until bone union is achieved, may lead to prolonged immobilization. The time to return to work may be longer.
- Stiffness, joint deformities, and contractures may be seen following surgeries, especially in the hands.
- Malignant transformation to chondrosarcoma may occur following skeletal maturity. However, transformation of a solitary enchondromatous lesion is rare (less than 1%).[40]
- Malignant transformation risk is increased in Ollier disease and Maffucci syndrome.[41][42] These two conditions are also associated with non-sarcomatous, extraosseous neoplasms, including brain tumors.[43]
Prompt consultation with an orthopedic specialist regarding this condition helps minimize complications.
Deterrence and Patient Education
Enchondroma-specific prevention measures are unavailable, as the cause of this condition is poorly understood. Genetic mutations are sporadic if present, so a clear inheritance pattern has not been elucidated. However, patients may be counseled about making healthy lifestyle choices to maintain bone health. Such measures include the following:
- Maintaining a healthy diet, particularly one incorporating rich calcium and vitamin D sources
- Having regular primary care visits, especially if the individual has a family history of bone disease
- Avoiding habits that may harm bone health, such as smoking and drinking
- Practicing safety in sports and recreation to prevent injuries
Patients must be reminded to seek prompt medical attention if they experience persistent musculoskeletal symptoms.
Pearls and Other Issues
The most important points to remember about enchondroma management are the following:
- Enchondroma is a benign hyaline cartilage tumor arising within a long bone's medullary cavity.
- Most enchondromatous lesions are solitary. Enchondromatosis is the occurrence of multiple enchondromas.
- The cause of the condition is unknown. Genetic mutations, if present, are sporadic.
- These lesions usually develop in small hand and foot bones, though they may also grow in the femur and humerus.
- Enchondromas increase the risk of pathologic fractures.
- These lesions may disrupt normal bone development in children.
- Enchondromas are often asymptomatic and discovered accidentally on imaging for another condition or if a pathologic fracture develops.
- Solitary lesions have a small chance of malignant transformation.
- Enchondromatosis, as in cases of Ollier disease and Maffucci syndrome, is associated with an increased risk of chondrosarcoma and visceral malignancies.
- Definitive diagnosis often involves a histological examination of the tissue, typically obtained through a biopsy or surgical tumor removal.
- Enchondromas typically appear as well-defined, radiolucent areas with small, stippled calcifications on x-rays. Other imaging modalities like CT and MRI may help determine other important tumor characteristics, like bone infarcts and aggressive features.
- Asymptomatic enchondromas do not require immediate treatment.
- Surgery may be considered in individuals with persistent symptoms or increased fracture risk and if concerns about malignant transformation are great.
Specific preventive measures for this condition are unavailable. However, prompt surgical intervention is beneficial for patients at risk for pathologic fractures or who have developed one.
Enhancing Healthcare Team Outcomes
Enchondromas are best managed with an interprofessional approach. The members of the multidisciplinary team should include the following:
- Primary care physicians: responsible for initial patient evaluation, referrals, and care coordination
- Radiologists: interpret imaging studies to assess the tumor's characteristics in aid of management
- Orthopedic surgeons: share their expertise in evaluating and treating individuals with suspected enchondroma. Orthopedic surgeons determine the need for surgical intervention and perform a biopsy or tumor excision if required.
- Pain management specialists: provide effective pain management to individuals experiencing pain associated with enchondromas. These professionals also provide anesthesia care if surgery is performed.
- Pathologists: analyze tissue samples obtained through biopsy or surgical excision to confirm the diagnosis and rule out malignancy
- Oncologists: share their expertise if concerns about malignant transformation are great or if a diagnosis of chondrosarcoma is confirmed. Oncologists also collaborate with orthopedic surgeons to develop a definitive treatment plan.
- Physical therapists: help with rehabilitation after surgical intervention, especially if joints are involved, or patients develop functional limitations
- Genetic counselors: may provide information about the genetic aspects of the condition and potential implications for family members
- Rehabilitation specialists: collaborate with physical therapists to optimize rehabilitation strategies. These professionals may also recommend assistive devices if necessary.
Effective communication and collaboration among these professionals are essential for providing holistic care, making accurate diagnoses, and tailoring treatment plans for individuals with enchondroma.
Media
(Click Image to Enlarge)
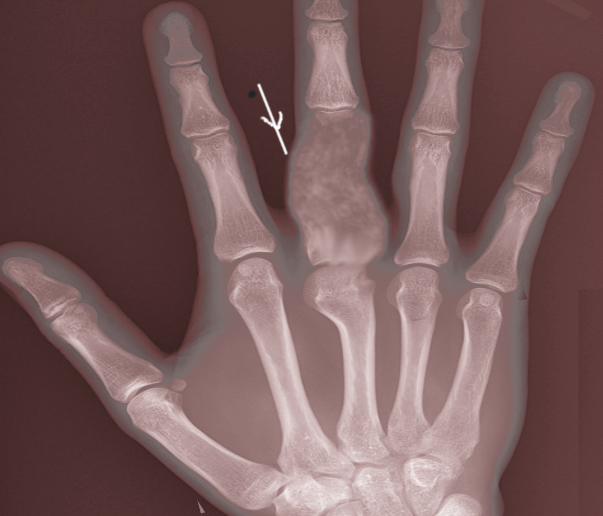
Enchondroma Radiography. This posteroanterior x-ray shows rarefaction mostly in the diaphysis and proximal metaphysis of the right 3rd proximal phalanx.
Image courtesy S Bhimji MD
References
Suster D, Hung YP, Nielsen GP. Differential Diagnosis of Cartilaginous Lesions of Bone. Archives of pathology & laboratory medicine. 2020 Jan:144(1):71-82. doi: 10.5858/arpa.2019-0441-RA. Epub [PubMed PMID: 31877083]
Simon MJ, Pogoda P, Hövelborn F, Krause M, Zustin J, Amling M, Barvencik F. Incidence, histopathologic analysis and distribution of tumours of the hand. BMC musculoskeletal disorders. 2014 May 28:15():182. doi: 10.1186/1471-2474-15-182. Epub 2014 May 28 [PubMed PMID: 24885007]
Level 2 (mid-level) evidenceSassoon AA, Fitz-Gibbon PD, Harmsen WS, Moran SL. Enchondromas of the hand: factors affecting recurrence, healing, motion, and malignant transformation. The Journal of hand surgery. 2012 Jun:37(6):1229-34. doi: 10.1016/j.jhsa.2012.03.019. Epub 2012 Apr 27 [PubMed PMID: 22542061]
Kerr DA, Cipriani NA. Benign Cartilage-forming Tumors. Surgical pathology clinics. 2021 Dec:14(4):585-603. doi: 10.1016/j.path.2021.06.004. Epub 2021 Oct 7 [PubMed PMID: 34742482]
Wessel LE, Christ AB, Athanasian EA. Impact of Patient and Tumor Characteristics on Range of Motion and Recurrence Following Treatment of Enchondromas of the Hand. The Journal of hand surgery. 2023 May:48(5):512.e1-512.e7. doi: 10.1016/j.jhsa.2021.11.027. Epub 2022 Jan 31 [PubMed PMID: 35115192]
Lubahn JD, Bachoura A. Enchondroma of the Hand: Evaluation and Management. The Journal of the American Academy of Orthopaedic Surgeons. 2016 Sep:24(9):625-33. doi: 10.5435/JAAOS-D-15-00452. Epub [PubMed PMID: 27454024]
Mulligan ME. How to Diagnose Enchondroma, Bone Infarct, and Chondrosarcoma. Current problems in diagnostic radiology. 2019 May-Jun:48(3):262-273. doi: 10.1067/j.cpradiol.2018.04.002. Epub 2018 Apr 6 [PubMed PMID: 29724496]
Zhang H, Alman BA. Enchondromatosis and Growth Plate Development. Current osteoporosis reports. 2021 Feb:19(1):40-49. doi: 10.1007/s11914-020-00639-7. Epub 2020 Dec 11 [PubMed PMID: 33306166]
Pansuriya TC, van Eijk R, d'Adamo P, van Ruler MA, Kuijjer ML, Oosting J, Cleton-Jansen AM, van Oosterwijk JG, Verbeke SL, Meijer D, van Wezel T, Nord KH, Sangiorgi L, Toker B, Liegl-Atzwanger B, San-Julian M, Sciot R, Limaye N, Kindblom LG, Daugaard S, Godfraind C, Boon LM, Vikkula M, Kurek KC, Szuhai K, French PJ, Bovée JV. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nature genetics. 2011 Nov 6:43(12):1256-61. doi: 10.1038/ng.1004. Epub 2011 Nov 6 [PubMed PMID: 22057234]
Level 2 (mid-level) evidenceAmary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F, McCarthy S, Fantin VR, Straley KS, Lobo S, Aston W, Green CL, Gale RE, Tirabosco R, Futreal A, Campbell P, Presneau N, Flanagan AM. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nature genetics. 2011 Nov 6:43(12):1262-5. doi: 10.1038/ng.994. Epub 2011 Nov 6 [PubMed PMID: 22057236]
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer cell. 2011 Jan 18:19(1):17-30. doi: 10.1016/j.ccr.2010.12.014. Epub [PubMed PMID: 21251613]
Level 3 (low-level) evidenceSuijker J, Baelde HJ, Roelofs H, Cleton-Jansen AM, Bovée JV. The oncometabolite D-2-hydroxyglutarate induced by mutant IDH1 or -2 blocks osteoblast differentiation in vitro and in vivo. Oncotarget. 2015 Jun 20:6(17):14832-42 [PubMed PMID: 26046462]
Deng Q, Li P, Che M, Liu J, Biswas S, Ma G, He L, Wei Z, Zhang Z, Yang Y, Liu H, Li B. Activation of hedgehog signaling in mesenchymal stem cells induces cartilage and bone tumor formation via Wnt/β-Catenin. eLife. 2019 Sep 4:8():. doi: 10.7554/eLife.50208. Epub 2019 Sep 4 [PubMed PMID: 31482846]
Satti LR, Yennapu NR, Inturi R, Surada R. A Rare Occurrence of Enchondroma in the Head of Femur in an Adult Male: A Case Report. Journal of orthopaedic case reports. 2023 Apr:13(4):62-65. doi: 10.13107/jocr.2023.v13.i04.3618. Epub [PubMed PMID: 37193382]
Level 3 (low-level) evidenceDing C, Chen W, Liu F, Xiong M, Chen J. Skull Base Chondrosarcoma Caused by Ollier Disease: A Case Report and Literature Review. World neurosurgery. 2019 Jul:127():103-108. doi: 10.1016/j.wneu.2019.03.037. Epub 2019 Mar 12 [PubMed PMID: 30872199]
Level 3 (low-level) evidenceProkopchuk O, Andres S, Becker K, Holzapfel K, Hartmann D, Friess H. Maffucci syndrome and neoplasms: a case report and review of the literature. BMC research notes. 2016 Feb 27:9():126. doi: 10.1186/s13104-016-1913-x. Epub 2016 Feb 27 [PubMed PMID: 26920730]
Level 3 (low-level) evidenceHakim DN, Pelly T, Kulendran M, Caris JA. Benign tumours of the bone: A review. Journal of bone oncology. 2015 Jun:4(2):37-41. doi: 10.1016/j.jbo.2015.02.001. Epub 2015 Mar 2 [PubMed PMID: 26579486]
Silve C, Jüppner H. Ollier disease. Orphanet journal of rare diseases. 2006 Sep 22:1():37 [PubMed PMID: 16995932]
Mittermayer F, Dominkus M, Krepler P, Schwameis E, Sluga M, Toma C, Lang S, Grampp S, Kotz R. Chondrosarcoma of the hand: is a wide surgical resection necessary? Clinical orthopaedics and related research. 2004 Jul:(424):211-5 [PubMed PMID: 15241167]
Lee L, Kazmer A, Colman MW, Gitelis S, Batus M, Blank AT. What is the clinical impact of staging and surveillance PET-CT scan findings in patients with bone and soft tissue sarcoma? Journal of surgical oncology. 2022 Apr:125(5):901-906. doi: 10.1002/jso.26789. Epub 2022 Jan 13 [PubMed PMID: 35023167]
Karaytuğ K, Alpan B, Bayram S, Valiyev N, Bilgiç B, Özger H. Long-term results of different surgical options in the management of solitary enchondroma. ANZ journal of surgery. 2022 Jul:92(7-8):1809-1813. doi: 10.1111/ans.17796. Epub 2022 May 27 [PubMed PMID: 35621280]
Bierry G, Kerr DA, Nielsen GP, Rosenberg AE, Huang AJ, Torriani M, Bredella MA. Enchondromas in children: imaging appearance with pathological correlation. Skeletal radiology. 2012 Sep:41(10):1223-9. doi: 10.1007/s00256-012-1377-6. Epub 2012 Feb 27 [PubMed PMID: 22366808]
Bachoura A, Rice IS, Lubahn AR, Lubahn JD. The surgical management of hand enchondroma without postcurettage void augmentation: authors' experience and a systematic review. Hand (New York, N.Y.). 2015 Sep:10(3):461-71. doi: 10.1007/s11552-015-9738-y. Epub [PubMed PMID: 26330779]
Level 1 (high-level) evidenceYildiz C, Erler K, Atesalp AS, Basbozkurt M. Benign bone tumors in children. Current opinion in pediatrics. 2003 Feb:15(1):58-67 [PubMed PMID: 12544273]
Level 3 (low-level) evidenceZhou X, Zhao B, Keshav P, Chen X, Gao W, Yan H. The management and surgical intervention timing of enchondromas: A 10-year experience. Medicine. 2017 Apr:96(16):e6678. doi: 10.1097/MD.0000000000006678. Epub [PubMed PMID: 28422880]
Patel S, Yadav S, Gurnani S, Yadav P, Selvin B. A Solitary Enchondroma of the Great Toe in an Adolescent Male: A Case Report. Cureus. 2022 Jan:14(1):e21772. doi: 10.7759/cureus.21772. Epub 2022 Jan 31 [PubMed PMID: 35251842]
Level 3 (low-level) evidenceTakigawa K. Chondroma of the bones of the hand. A review of 110 cases. The Journal of bone and joint surgery. American volume. 1971 Dec:53(8):1591-600 [PubMed PMID: 5121800]
Level 3 (low-level) evidenceDavies AM, Patel A, Azzopardi C, James SL, Botchu R. Prevalence of Enchondromas of the Proximal Femur in Adults as an Incidental Finding on MRI of the Pelvis. The Indian journal of radiology & imaging. 2021 Jul:31(3):582-585. doi: 10.1055/s-0041-1735915. Epub 2021 Oct 6 [PubMed PMID: 34790301]
Sullivan CW, Kazley JM, Murtaza H, Cooley M, Jones D, DiCaprio MR. Team Approach: Evaluation and Management of Low-Grade Cartilaginous Lesions. JBJS reviews. 2020 Jan:8(1):e0054. doi: 10.2106/JBJS.RVW.19.00054. Epub [PubMed PMID: 32105237]
Jeong W, Kim HJ. Biomarkers of chondrosarcoma. Journal of clinical pathology. 2018 Jul:71(7):579-583. doi: 10.1136/jclinpath-2018-205071. Epub 2018 Mar 28 [PubMed PMID: 29593061]
Georgiannos D, Lampridis V, Bisbinas I. Phenolization and coralline hydroxyapatite grafting following meticulous curettage for the treatment of enchondroma of the hand. A case series of 82 patients with 5-year follow-up. Hand (New York, N.Y.). 2015 Mar:10(1):111-5. doi: 10.1007/s11552-014-9674-2. Epub [PubMed PMID: 25767429]
Level 2 (mid-level) evidenceBickels J, Wittig JC, Kollender Y, Kellar-Graney K, Mansour KL, Meller I, Malawer MM. Enchondromas of the hand: treatment with curettage and cemented internal fixation. The Journal of hand surgery. 2002 Sep:27(5):870-5 [PubMed PMID: 12239678]
Howard EL, Shepherd KL, Cribb G, Cool P. The validity of the Mirels score for predicting impending pathological fractures of the lower limb. The bone & joint journal. 2018 Aug:100-B(8):1100-1105. doi: 10.1302/0301-620X.100B8.BJJ-2018-0300.R1. Epub [PubMed PMID: 30062934]
Leung CH, Lui TH. Endoscopic Curettage and Bone Grafting of Enchondroma of Proximal Phalanx of Finger. Arthroscopy techniques. 2023 Aug:12(8):e1335-e1340. doi: 10.1016/j.eats.2023.04.001. Epub 2023 Jul 10 [PubMed PMID: 37654891]
Kaempf de Oliveira R, Brunelli JPF, Araújo Filho R, Aita MA, Delgado PJ. Intramedullary Fixation with Cannulated Screw After Resection of Enchondroma in the Hand: Technique Description and Case Series. Journal of hand surgery global online. 2023 Jul:5(4):413-420. doi: 10.1016/j.jhsg.2023.03.008. Epub 2023 Apr 6 [PubMed PMID: 37521559]
Level 2 (mid-level) evidenceFutani H, Kawaguchi T, Sawai T, Tachibana T. Osteoscopic versus open surgery for the treatment of enchondroma in the foot. Archives of orthopaedic and trauma surgery. 2023 Aug:143(8):4899-4905. doi: 10.1007/s00402-023-04816-y. Epub 2023 Feb 23 [PubMed PMID: 36813947]
Yiannakas M, Ioannides C, Pantzara M, Michaelides M. Radiofrequency ablation for the treatment of a presumed enchondroma in the flat bones of the pelvis. Skeletal radiology. 2023 May:52(5):1057-1061. doi: 10.1007/s00256-023-04291-x. Epub 2023 Feb 11 [PubMed PMID: 36773084]
Baeza-Trinidad R, Oteo Revuelta JA. Images in clinical medicine. Spina ventosa. The New England journal of medicine. 2015 Mar 26:372(13):e18. doi: 10.1056/NEJMicm1315601. Epub [PubMed PMID: 25806937]
Level 3 (low-level) evidenceYoshida T, Sakamoto A, Tanaka K, Matsuda S, Oda Y, Iwamoto Y. Alternative surgical treatment for giant-cell reparative granuloma in the metacarpal, using phenol and ethanol adjuvant therapy. The Journal of hand surgery. 2007 Jul-Aug:32(6):887-92 [PubMed PMID: 17606072]
Level 3 (low-level) evidenceAltay M, Bayrakci K, Yildiz Y, Erekul S, Saglik Y. Secondary chondrosarcoma in cartilage bone tumors: report of 32 patients. Journal of orthopaedic science : official journal of the Japanese Orthopaedic Association. 2007 Sep:12(5):415-23 [PubMed PMID: 17909925]
Level 2 (mid-level) evidenceSchwartz HS, Zimmerman NB, Simon MA, Wroble RR, Millar EA, Bonfiglio M. The malignant potential of enchondromatosis. The Journal of bone and joint surgery. American volume. 1987 Feb:69(2):269-74 [PubMed PMID: 3805090]
Albregts AE, Rapini RP. Malignancy in Maffucci's syndrome. Dermatologic clinics. 1995 Jan:13(1):73-8 [PubMed PMID: 7712654]
Lewis RJ, Ketcham AS. Maffucci's syndrome: functional and neoplastic significance. Case report and review of the literature. The Journal of bone and joint surgery. American volume. 1973 Oct:55(7):1465-79 [PubMed PMID: 4586088]
Level 3 (low-level) evidence