Introduction
Apert syndrome, also known as acrocephalosyndactyly type I, is a genetically inherited syndrome characterized by multisuture craniosynostosis, midface retrusion, and syndactyly. The syndrome was first described in 1906 by French physician Eugene Apert, who described 9 people with similar facial and extremity characteristics.[1]
The condition is caused by mutations in the FGFR2 gene, which encodes a protein that regulates cell and bone growth, crucial for normal skull, face, and limb formation. The genetic anomalies lead to abnormal bone development. Advanced paternal age is a significant risk factor for de novo mutations in Apert syndrome. Diagnosis is based on clinical features, supported by genetic testing for FGFR2 mutations. Treatment often involves surgical correction of craniosynostosis and syndactyly, alongside supportive therapies. With early intervention, affected individuals can have a near-normal life expectancy, though they may experience developmental and neurological challenges.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Apert syndrome is inherited in an autosomal dominant fashion, though most cases result from de novo mutations. The condition is commonly caused by a gain-of-function missense mutation in exon IIIa of the FGFR2 gene on chromosome 10q, leading to amino acid substitutions Ser252Trp or Pro253Arg.[2][3]. Phenotypic differences may arise depending on the mutation. Patients with the Pro253Arg mutation often have more severe syndactyly, while the Ser252Trp mutation is associated with cleft palate.[4][5]
Epidemiology
Apert syndrome is a rare disease, estimated to occur in 1 in 65,000 to 200,000 births, depending on the study cited.[6] Male and female infants are equally affected. The incidence of the disease significantly increases with paternal age and is thought to provide a selective advantage within male spermatogonial cells.[7] The syndrome exhibits complete penetrance with variable expressivity, resulting in a spectrum of clinical presentations, from subtle findings to severe deformities, even within the same family. The condition has been reported to occur 2.9 times more frequently in the Asian population compared to Hispanics.[8]
Pathophysiology
Two-thirds of cases of Apert syndrome are due to a specific cytosine-to-guanine mutation at position 755 of the FGFR2 gene, resulting in a serine-to-tryptophan amino acid change on the paternally derived allele.[9] Several mouse models have been developed that provide insight into the underlying pathophysiology of these mutations. In mice, the FGFR2 receptor loses its specificity and can bind other fibroblast growth factors, suppressing apoptosis of osteoblasts, which leads to syndactyly and craniosynostosis. The exact underlying mechanism remains unclear but is likely linked to a specific fibroblast growth factor.[10][11]
History and Physical
The family history of patients suspected of having Apert syndrome is crucial to evaluation due to its autosomal dominant inheritance. While a lack of family history does not rule out the diagnosis because of the possibility of de novo mutations, a positive family history significantly increases its likelihood.
Patients with Apert syndrome typically present with craniosynostosis, midface hypoplasia, and symmetric syndactyly of the hands and feet (see Image. Cranioacial and Oral Findings in Apert Syndrome). The craniosynostosis is more severe compared to that seen in Crouzon syndrome, and the presence of syndactyly helps confirm the diagnosis. However, features such as hypertelorism (wide-set eyes), proptosis (bulging eyes), and down-slanting palpebral fissures are common in several craniosynostosis syndromes and cannot differentiate these conditions. Most patients also have a large anterior fontanelle displaced anteriorly. The midface is usually underdeveloped and retruded, leading to the underdevelopment of shallow orbits and down-slanting palpebral fissures.[12]
In addition to down-slanting palpebral fissures, patients with Apert syndrome are at risk for other ocular abnormalities, including strabismus, refractive errors, and anisometropia.[13][14] Hearing loss occurs in up to 80% of affected individuals, typically the conductive type, due to otitis media with effusion, ossicular abnormalities, and stenosis of the external auditory canal.[15][16][17]
Many patients with Apert syndrome experience multilevel airway obstruction due to narrow nasal passages, tongue-based airway obstruction, or tracheal anomalies.[18] Obstructive sleep apnea is also common, affecting roughly 30% of patients, and can persist despite midface advancement.[19][20]
Hand abnormalities are characterized by a short, radially deviated thumb, complex syndactyly of the middle 3 digits, syndactyly of the 4th webspace, and symphalangism or the congenital stiffness of the fingers due to failure of bone separation during fetal growth. Three specific subtypes of hand findings in Apert syndrome are identified based on hand shape: spade (side-to-side fusion with a flat palm), mitten (fusion of fingers resulting in a concave palm), and rosebud (tight fusion of all digits).[21] The fusion of the fingernails of the 2nd to 4th digits to form a single nail is referred to as "synonychia" (see Image. Hand Findings in Apert Syndrome).
Other craniofacial deformities in Apert syndrome include acrocephaly (cone-shaped calvarium), proptosis, prominent forehead, hypertelorism, down-slanting palpebral fissures, and a flattened nasal bridge. Oral findings include dental crowding, high-arched palate, narrow palate, and pseudo-clefts. Skeletal abnormalities may include cervical vertebrae fusions, often involving C5-C6, scoliosis, atlantoaxial subluxation, or C1 spina bifida occulta.[22]
Neurological involvement in Apert syndrome typically manifests as nonprogressive ventriculomegaly, corpus callosum abnormalities, jugular foramen stenosis, absent septum pellucidum, Chiari malformations, posterior fossa arachnoid cysts, and limbic defects.[23][24][25] While most patients with Apert syndrome have normal cognition or mild intellectual impairment, some have been reported to experience moderate-to-severe disability.[26][27]
Visceral anomalies associated with Apert syndrome include cardiovascular defects such as ventricular septal defects and overriding aortas, intestinal malrotation, distal esophagus stenosis, and pyloric stenosis. Genitourinary anomalies may include hydronephrosis or cryptorchidism.[28]
Evaluation
The evaluation of Apert syndrome in a patient with a known family history is primarily clinical, as the characteristic physical findings confirm the diagnosis. Additional testing is required in cases where the clinical presentation is unclear and no family history is present. Brain magnetic resonance imaging (MRI) and computed tomography (CT) help detect craniosynostosis or other skeletal abnormalities, including perisutural sclerosis, reduced serration, bony bridging, and the absence of sutures. The coronal sutures are most commonly affected, with variable involvement of the sagittal and lambdoid sutures. These imaging techniques also detect other abnormalities, such as increased intracranial pressure (ICP) and Chiari malformations.
ICP monitoring may be necessary due to the risk of intracranial hypertension, especially in syndromic craniosynostosis.[29][30] The ICP rises due to hydrocephalus, osseous changes in the skull base affecting venous outflow, and midface hypoplasia, leading to sleep apnea. Screening for intracranial hypertension begins with an ophthalmological assessment to check for papilledema. Optical coherence tomography is also effective for detecting ICP elevation.[31][32] ICP monitoring is indicated if these methods are inconclusive.[33][34] Polysomnography should be obtained to evaluate for sleep apnea in patients with suspected sleep-disordered breathing.[35][36]
Genetic and molecular testing can be pursued, as with other craniosynostosis syndromes where the diagnosis is unclear or features are atypical. The underlying mechanism of many craniosynostosis syndromes involves FGFR mutations and abnormal signaling. Prenatal genetic testing, MRI, and ultrasound can help confirm the diagnosis before birth.[37][38] Although amniocentesis and chorionic villus sampling are options, safer imaging techniques are likely to replace these higher-risk procedures, except in challenging cases.
As mentioned, history, physical examination, and imaging findings are used to confirm the specific craniosynostosis diagnosis. However, distinguishing between syndromes, including Pfeiffer, Apert, Saethre Chotzen, Carpenter, and Jackson Weiss, can be challenging due to significant overlap.
Treatment / Management
Like other craniosynostoses, management requires a team-based approach with multiple subspecialists, including pediatricians, neurosurgeons, plastic surgeons, craniofacial surgeons, ophthalmologists, and dentists. Surgery is necessary to prevent complications related to intracranial hypertension and protect the developing brain.
Several surgical approaches for craniosynostosis correction have been described in the literature, including fronto-orbital advancements, posterior vault distractions, and endoscopic strip craniectomies.[39][40][41][42][43][44][45] Surgeries for midface retrusion correction include Le Fort and monobloc distractions.[46][47][48][49][50] A detailed description of the surgical management of syndromic craniosynostosis can be found in the updated guidelines on the treatment and management of craniosynostosis.[51]
If hydrocephalus is present, treatment may involve placing a shunt or endoscopic 3rd ventriculostomy with or without choroid plexus coagulation.[52] When placing a ventriculoperitoneal shunt, the need for future open vault reconstructions must be considered. Placing a frontal shunt may expose the shunt hardware during vault reconstruction. A parieto-occipital shunt may be a better option. If concerns about future operations arise, an endoscopic 3rd ventriculostomy may be a reasonable first approach.
De Jong and colleagues published a treatment protocol for patients with syndromic craniosynostosis, recommending fundoscopy and polysomnography annually until 6 years of age, as well as hearing evaluations until 4 years of age. Pure tone audiometry is indicated if hearing evaluations are abnormal.
Differential Diagnosis
Apert syndrome shares overlapping features with several craniosynostosis syndromes. Consideration of these conditions, including the disorders below, is critical to avoid misdiagnosis and ensure targeted treatment.
- Achondroplasia
- Antley Bixler syndrome
- Beare Stevenson syndrome
- Crouzon syndrome
- Cutis gyrata
- Pfeiffer syndrome
- Thanatophoric dysplasia
- Muenke syndrome
- Jackson Weiss syndrome
- Saethre Chotzen syndrome
Timely and accurate diagnosis can significantly impact patient outcomes and guide the management of associated complications. Recognizing the differential diagnoses of Apert syndrome ensures that clinicians provide the most appropriate treatment plan.
Prognosis
As with many congenital conditions, early prenatal and perinatal diagnosis is crucial for counseling families regarding prognosis. Factors influencing mental development include the age at the first surgery, associated brain malformations, and family environment quality. One study found that 32% of patients had an intelligence quotient of at least 70, with over 50% having an intelligence quotient greater than 70 if surgery occurred before age 1 year.[53] Long-term follow-up studies indicate that patients often have positive psychosocial outcomes.[54][55]
Continued follow-up is vital to minimize complications related to craniosynostosis, such as strabismus, sleep apnea, and intracranial hypertension. These issues do not always fully resolve after surgery for individuals with cranial and facial defects. In a retrospective Australian study, 54% develop vision loss in at least 1 eye due to amblyopia after craniofacial surgery for Apert syndrome. Fortunately, optic atrophy was found in only 5% of cases, likely due to the widespread use of early craniofacial surgery for craniosynostosis syndromes.[56] Strabismus is also highly prevalent, developing in 2/3 of patients.[57] Severe-to-profound hearing loss is also more common in syndromic craniosynostoses than in nonsyndromic variants.[58] A team-based approach involving multiple subspecialists is essential for monitoring vision and life-threatening complications.
Complications
The main complications of Apert syndrome include increased ICP, which can lead to papilledema and cognitive impairment, as well as exposure keratopathy and corneal scarring. Respiratory issues, particularly due to sleep apnea, are common, and spinal cord injuries or neurologic deficits may occur in patients with cervical spine anomalies. Additionally, aspiration pneumonia and chronic lung disease are concerns that may develop. Managing these complications requires careful monitoring and an interprofessional approach to address the various health challenges associated with the syndrome.
Consultations
Given the complexity of Apert syndrome, patients require care from a variety of specialties to address the multifaceted challenges of the condition. Consultations must include the following:
- Neurosurgery
- Ophthalmology (pediatric, oculoplastic)
- Plastic surgery
- Maxillofacial surgery
- Otorhinolaryngology
- Audiology
- Dentistry
- Orthodontics
- Geneticist
An interprofessional approach is essential for providing comprehensive care to patients with Apert syndrome. Such collaboration improves patient outcomes and minimizes long-term complications.
Deterrence and Patient Education
Apert syndrome follows an autosomal dominant inheritance pattern, with advanced paternal age associated with de novo occurrences. The chance of passing the genetic trait to each child is 50%. If a pathogenic variant is present in the family, prenatal testing should be offered for pregnancies at increased risk.
Enhancing Healthcare Team Outcomes
Like other craniosynostoses, the management of Apert syndrome involves a team-based approach with multiple subspecialists, including pediatricians, neurosurgeons, plastic surgeons, craniofacial surgeons, ophthalmologists, and dentists. Surgical intervention is essential to prevent complete closure of the coronal suture and safeguard brain development. Collaboration among subspecialists is also essential to address complications such as strabismus, sleep apnea, and intracranial hypertension. An interprofessional team plays a critical role in monitoring for vision and life-threatening complications, making informed surgical decisions, and ensuring optimal long-term outcomes.
Media
(Click Image to Enlarge)
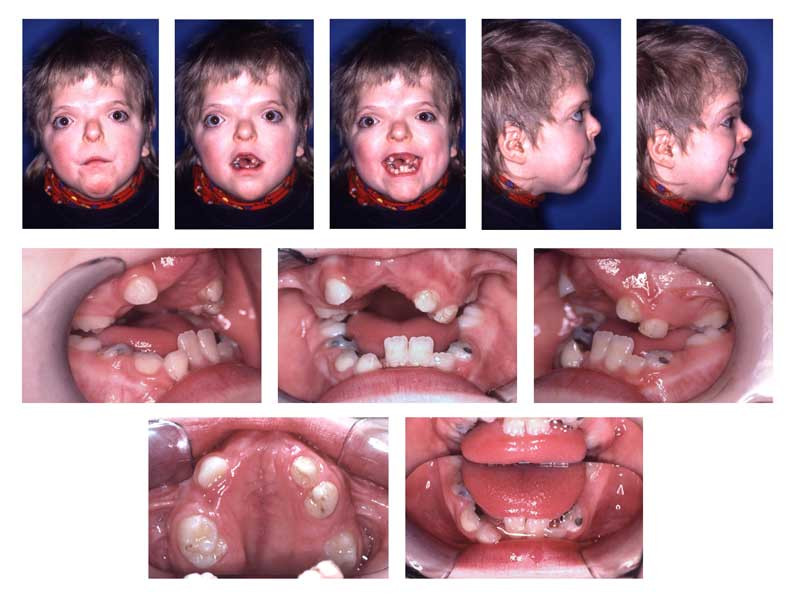
Craniofacial and Oral Findings in Apert Syndrome. The image depicts hypertelorism, vertical excess of the lower one-third of the face, a trapezoidal upper lip, and difficulty achieving forced lip closure. Intraoral changes are delayed dentition, crowding, Angle Class III malocclusion with dental compensation through lower incisor retrusion, and a circular open bite with a unilateral crossbite.
Filip, Public Domain, via Wikimedia Commons
{kind=link}
(Click Image to Enlarge)
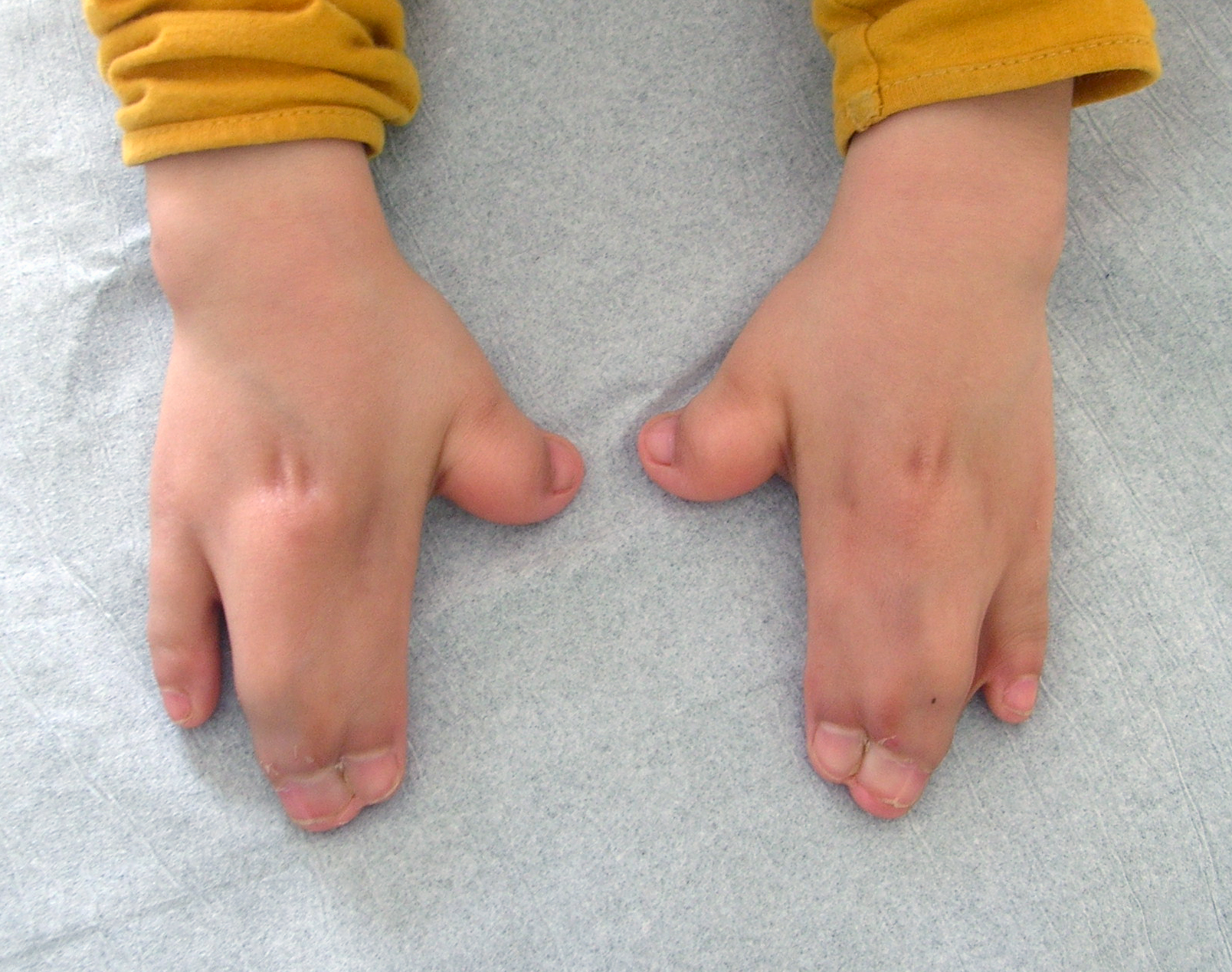
Hand Findings in Apert Syndrome. This image shows the characteristic fusion of the fingers (syndactyly) and nails (synonychia) commonly seen in individuals with Apert syndrome. These findings reflect the underlying genetic disruption in limb development associated with the condition.
Gzzz, Public Domain, via Wikimedia Commons
References
Kutkowska-Kaźmierczak A, Gos M, Obersztyn E. Craniosynostosis as a clinical and diagnostic problem: molecular pathology and genetic counseling. Journal of applied genetics. 2018 May:59(2):133-147. doi: 10.1007/s13353-017-0423-4. Epub 2018 Feb 1 [PubMed PMID: 29392564]
Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nature genetics. 1995 Feb:9(2):165-72 [PubMed PMID: 7719344]
Das S, Munshi A. Research advances in Apert syndrome. Journal of oral biology and craniofacial research. 2018 Sep-Dec:8(3):194-199. doi: 10.1016/j.jobcr.2017.05.006. Epub 2017 May 25 [PubMed PMID: 30191107]
Level 3 (low-level) evidenceSlaney SF, Oldridge M, Hurst JA, Moriss-Kay GM, Hall CM, Poole MD, Wilkie AO. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. American journal of human genetics. 1996 May:58(5):923-32 [PubMed PMID: 8651276]
von Gernet S, Golla A, Ehrenfels Y, Schuffenhauer S, Fairley JD. Genotype-phenotype analysis in Apert syndrome suggests opposite effects of the two recurrent mutations on syndactyly and outcome of craniofacial surgery. Clinical genetics. 2000 Feb:57(2):137-9 [PubMed PMID: 10735635]
Fearon JA. Treatment of the hands and feet in Apert syndrome: an evolution in management. Plastic and reconstructive surgery. 2003 Jul:112(1):1-12; discussion 13-9 [PubMed PMID: 12832871]
Level 2 (mid-level) evidenceGoriely A, McVean GA, Röjmyr M, Ingemarsson B, Wilkie AO. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science (New York, N.Y.). 2003 Aug 1:301(5633):643-6 [PubMed PMID: 12893942]
Tolarova MM, Harris JA, Ordway DE, Vargervik K. Birth prevalence, mutation rate, sex ratio, parents' age, and ethnicity in Apert syndrome. American journal of medical genetics. 1997 Nov 12:72(4):394-8 [PubMed PMID: 9375719]
Moloney DM, Slaney SF, Oldridge M, Wall SA, Sahlin P, Stenman G, Wilkie AO. Exclusive paternal origin of new mutations in Apert syndrome. Nature genetics. 1996 May:13(1):48-53 [PubMed PMID: 8673103]
Hajihosseini MK, Duarte R, Pegrum J, Donjacour A, Lana-Elola E, Rice DP, Sharpe J, Dickson C. Evidence that Fgf10 contributes to the skeletal and visceral defects of an Apert syndrome mouse model. Developmental dynamics : an official publication of the American Association of Anatomists. 2009 Feb:238(2):376-85. doi: 10.1002/dvdy.21648. Epub [PubMed PMID: 18773495]
Level 3 (low-level) evidenceYapijakis C, Pachis N, Sotiriadou T, Vaila C, Michopoulou V, Vassiliou S. Molecular Mechanisms Involved in Craniosynostosis. In vivo (Athens, Greece). 2023 Jan-Feb:37(1):36-46. doi: 10.21873/invivo.13052. Epub [PubMed PMID: 36593018]
Cohen MM Jr, Kreiborg S. A clinical study of the craniofacial features in Apert syndrome. International journal of oral and maxillofacial surgery. 1996 Feb:25(1):45-53 [PubMed PMID: 8833300]
Khong JJ, Anderson P, Gray TL, Hammerton M, Selva D, David D. Ophthalmic findings in apert syndrome prior to craniofacial surgery. American journal of ophthalmology. 2006 Aug:142(2):328-30 [PubMed PMID: 16876521]
Rostamzad P, Arslan ZF, Mathijssen IMJ, Koudstaal MJ, Pleumeekers MM, Versnel SL, Loudon SE. Prevalence of Ocular Anomalies in Craniosynostosis: A Systematic Review and Meta-Analysis. Journal of clinical medicine. 2022 Feb 18:11(4):. doi: 10.3390/jcm11041060. Epub 2022 Feb 18 [PubMed PMID: 35207332]
Level 1 (high-level) evidenceAgochukwu NB, Solomon BD, Muenke M. Hearing loss in syndromic craniosynostoses: otologic manifestations and clinical findings. International journal of pediatric otorhinolaryngology. 2014 Dec:78(12):2037-47. doi: 10.1016/j.ijporl.2014.09.019. Epub 2014 Sep 28 [PubMed PMID: 25441602]
Agochukwu NB, Solomon BD, Muenke M. Impact of genetics on the diagnosis and clinical management of syndromic craniosynostoses. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2012 Sep:28(9):1447-63. doi: 10.1007/s00381-012-1756-2. Epub 2012 Aug 8 [PubMed PMID: 22872262]
Rajenderkumar D, Bamiou DE, Sirimanna T. Audiological profile in Apert syndrome. Archives of disease in childhood. 2005 Jun:90(6):592-3 [PubMed PMID: 15908623]
Wenger TL, Dahl J, Bhoj EJ, Rosen A, McDonald-McGinn D, Zackai E, Jacobs I, Heike CL, Hing A, Santani A, Inglis AF, Sie KC, Cunningham M, Perkins J. Tracheal cartilaginous sleeves in children with syndromic craniosynostosis. Genetics in medicine : official journal of the American College of Medical Genetics. 2017 Jan:19(1):62-68. doi: 10.1038/gim.2016.60. Epub 2016 May 26 [PubMed PMID: 27228464]
de Jong T, Bannink N, Bredero-Boelhouwer HH, van Veelen ML, Bartels MC, Hoeve LJ, Hoogeboom AJ, Wolvius EB, Lequin MH, van der Meulen JJ, van Adrichem LN, Vaandrager JM, Ongkosuwito EM, Joosten KF, Mathijssen IM. Long-term functional outcome in 167 patients with syndromic craniosynostosis; defining a syndrome-specific risk profile. Journal of plastic, reconstructive & aesthetic surgery : JPRAS. 2010 Oct:63(10):1635-41. doi: 10.1016/j.bjps.2009.10.029. Epub 2009 Nov 12 [PubMed PMID: 19913472]
Bannink N, Nout E, Wolvius EB, Hoeve HL, Joosten KF, Mathijssen IM. Obstructive sleep apnea in children with syndromic craniosynostosis: long-term respiratory outcome of midface advancement. International journal of oral and maxillofacial surgery. 2010 Feb:39(2):115-21. doi: 10.1016/j.ijom.2009.11.021. Epub 2010 Jan 6 [PubMed PMID: 20056390]
Arroyo Carrera I, Martínez-Frías ML, Marco Pérez JJ, Paisán Grisolía L, Cárdenes Rodríguez A, Nieto Conde C, Félix Rodríguez V, Egüés Jimeno JJ, Morales Fernández MC, Gómez-Ullate Vergara J, Pardo Romero M, Peñas Valiente A, Oliván del Cacho MJ, Lara Palma A. [Apert syndrome: clinico-epidemiological analysis of a series of consecutive cases in Spain]. Anales espanoles de pediatria. 1999 Dec:51(6):667-72 [PubMed PMID: 10666902]
Level 2 (mid-level) evidenceBreik O, Mahindu A, Moore MH, Molloy CJ, Santoreneos S, David DJ. Central nervous system and cervical spine abnormalities in Apert syndrome. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2016 May:32(5):833-8. doi: 10.1007/s00381-016-3036-z. Epub 2016 Feb 10 [PubMed PMID: 26861132]
Cinalli G, Renier D, Sebag G, Sainte-Rose C, Arnaud E, Pierre-Kahn A. Chronic tonsillar herniation in Crouzon's and Apert's syndromes: the role of premature synostosis of the lambdoid suture. Journal of neurosurgery. 1995 Oct:83(4):575-82 [PubMed PMID: 7674004]
Quintero-Rivera F, Robson CD, Reiss RE, Levine D, Benson CB, Mulliken JB, Kimonis VE. Intracranial anomalies detected by imaging studies in 30 patients with Apert syndrome. American journal of medical genetics. Part A. 2006 Jun 15:140(12):1337-8 [PubMed PMID: 16691624]
Tan AP, Mankad K. Apert syndrome: magnetic resonance imaging (MRI) of associated intracranial anomalies. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2018 Feb:34(2):205-216. doi: 10.1007/s00381-017-3670-0. Epub 2017 Dec 2 [PubMed PMID: 29198073]
Fernandes MB, Maximino LP, Perosa GB, Abramides DV, Passos-Bueno MR, Yacubian-Fernandes A. Apert and Crouzon syndromes-Cognitive development, brain abnormalities, and molecular aspects. American journal of medical genetics. Part A. 2016 Jun:170(6):1532-7. doi: 10.1002/ajmg.a.37640. Epub 2016 Mar 30 [PubMed PMID: 27028366]
David DJ, Anderson P, Flapper W, Syme-Grant J, Santoreneos S, Moore M. Apert Syndrome: Outcomes From the Australian Craniofacial Unit's Birth to Maturity Management Protocol. The Journal of craniofacial surgery. 2016 Jul:27(5):1125-34. doi: 10.1097/SCS.0000000000002709. Epub [PubMed PMID: 27380568]
Cohen MM Jr, Kreiborg S. Visceral anomalies in the Apert syndrome. American journal of medical genetics. 1993 Mar 15:45(6):758-60 [PubMed PMID: 8456856]
Abu-Sittah GS, Jeelani O, Dunaway D, Hayward R. Raised intracranial pressure in Crouzon syndrome: incidence, causes, and management. Journal of neurosurgery. Pediatrics. 2016 Apr:17(4):469-75. doi: 10.3171/2015.6.PEDS15177. Epub 2015 Nov 27 [PubMed PMID: 26613275]
Marucci DD, Dunaway DJ, Jones BM, Hayward RD. Raised intracranial pressure in Apert syndrome. Plastic and reconstructive surgery. 2008 Oct:122(4):1162-1168. doi: 10.1097/PRS.0b013e31818458f0. Epub [PubMed PMID: 18827651]
Driessen C, Eveleens J, Bleyen I, van Veelen ML, Joosten K, Mathijssen I. Optical coherence tomography: a quantitative tool to screen for papilledema in craniosynostosis. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2014 Jun:30(6):1067-73. doi: 10.1007/s00381-014-2376-9. Epub 2014 Feb 12 [PubMed PMID: 24519451]
Swanson JW, Aleman TS, Xu W, Ying GS, Pan W, Liu GT, Lang SS, Heuer GG, Storm PB, Bartlett SP, Katowitz WR, Taylor JA. Evaluation of Optical Coherence Tomography to Detect Elevated Intracranial Pressure in Children. JAMA ophthalmology. 2017 Apr 1:135(4):320-328. doi: 10.1001/jamaophthalmol.2017.0025. Epub [PubMed PMID: 28241164]
Tamburrini G, Caldarelli M, Massimi L, Santini P, Di Rocco C. Intracranial pressure monitoring in children with single suture and complex craniosynostosis: a review. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2005 Oct:21(10):913-21 [PubMed PMID: 15871027]
Spruijt B, Joosten KFM, Driessen C, Rizopoulos D, Naus NC, van der Schroeff MP, Wolvius EB, van Veelen MC, Tasker RC, Mathijssen IMJ. Algorithm for the Management of Intracranial Hypertension in Children with Syndromic Craniosynostosis. Plastic and reconstructive surgery. 2015 Aug:136(2):331-340. doi: 10.1097/PRS.0000000000001434. Epub [PubMed PMID: 25909300]
Al-Saleh S, Riekstins A, Forrest CR, Philips JH, Gibbons J, Narang I. Sleep-related disordered breathing in children with syndromic craniosynostosis. Journal of cranio-maxillo-facial surgery : official publication of the European Association for Cranio-Maxillo-Facial Surgery. 2011 Apr:39(3):153-7. doi: 10.1016/j.jcms.2010.04.011. Epub 2010 Jun 2 [PubMed PMID: 20627744]
Tan HL, Kheirandish-Gozal L, Abel F, Gozal D. Craniofacial syndromes and sleep-related breathing disorders. Sleep medicine reviews. 2016 Jun:27():74-88. doi: 10.1016/j.smrv.2015.05.010. Epub 2015 Jun 6 [PubMed PMID: 26454241]
Varlas VN, Epistatu D, Varlas RG. Emphasis on Early Prenatal Diagnosis and Perinatal Outcomes Analysis of Apert Syndrome. Diagnostics (Basel, Switzerland). 2024 Jul 10:14(14):. doi: 10.3390/diagnostics14141480. Epub 2024 Jul 10 [PubMed PMID: 39061616]
Azoury SC, Reddy S, Shukla V, Deng CX. Fibroblast Growth Factor Receptor 2 (FGFR2) Mutation Related Syndromic Craniosynostosis. International journal of biological sciences. 2017:13(12):1479-1488. doi: 10.7150/ijbs.22373. Epub 2017 Nov 2 [PubMed PMID: 29230096]
Riesel JN, Riordan CP, Hughes CD, Karsten MB, Staffa SJ, Meara JG, Proctor MR. Endoscopic strip craniectomy with orthotic helmeting for safe improvement of head growth in children with Apert syndrome. Journal of neurosurgery. Pediatrics. 2022 Jun 1:29(6):659-666. doi: 10.3171/2022.2.PEDS21340. Epub 2022 Apr 1 [PubMed PMID: 35364592]
Yildizdal S, Kaplan GO, Akca B, Kucukguven A, Işikay I, Vargel I. Fronto-orbital advancement: Comparison of syndromic and nonsyndromic craniosynostosis patients. Journal of cranio-maxillo-facial surgery : official publication of the European Association for Cranio-Maxillo-Facial Surgery. 2025 Apr:53(4):420-427. doi: 10.1016/j.jcms.2025.01.015. Epub 2025 Jan 23 [PubMed PMID: 39848875]
Raposo-Amaral CE, Vincenzi-Lemes M, Medeiros ML, Raposo-Amaral CA, Ghizoni E. Apert syndrome: neurosurgical outcomes and complications following posterior vault distraction osteogenesis. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2024 Aug:40(8):2557-2563. doi: 10.1007/s00381-024-06436-2. Epub 2024 May 3 [PubMed PMID: 38700706]
de Jong T, van Veelen ML, Mathijssen IM. Spring-assisted posterior vault expansion in multisuture craniosynostosis. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2013 May:29(5):815-20. doi: 10.1007/s00381-013-2033-8. Epub 2013 Jan 26 [PubMed PMID: 23354443]
Breakey RWF, Mercan E, van de Lande LS, Sidpra J, Birgfeld C, Lee A, Schievano S, Dunaway DJ, Jeelani NO, Hopper RA. Two-Center Review of Posterior Vault Expansion following a Staged or Expectant Treatment of Crouzon and Apert Craniosynostosis. Plastic and reconstructive surgery. 2023 Mar 1:151(3):615-626. doi: 10.1097/PRS.0000000000009925. Epub 2022 Nov 22 [PubMed PMID: 36730425]
Lo WB, Thant KZ, Kaderbhai J, White N, Nishikawa H, Dover MS, Evans M, Rodrigues D. Posterior calvarial distraction for complex craniosynostosis and cerebellar tonsillar herniation. Journal of neurosurgery. Pediatrics. 2020 Oct 1:26(4):421-430. doi: 10.3171/2020.4.PEDS19742. Epub 2020 Jul 10 [PubMed PMID: 32650306]
Jimenez DF, Barone CM. Bilateral endoscopic craniectomies in the treatment of an infant with Apert syndrome. Journal of neurosurgery. Pediatrics. 2012 Oct:10(4):310-4. doi: 10.3171/2012.7.PEDS11281. Epub 2012 Aug 24 [PubMed PMID: 22920294]
Raposo-Amaral CE, Vieira PH, Denadai R, Ghizoni E, Raposo-Amaral CA. Treating Syndromic Craniosynostosis with Monobloc Facial Bipartition and Internal Distractor Devices: Destigmatizing the Syndromic Face. Clinics in plastic surgery. 2021 Jul:48(3):521-529. doi: 10.1016/j.cps.2021.03.002. Epub 2021 May 11 [PubMed PMID: 34051903]
Dunaway DJ, Budden C, Ong J, James G, Jeelani NUO. Monobloc Distraction and Facial Bipartition Distraction with External Devices. Clinics in plastic surgery. 2021 Jul:48(3):507-519. doi: 10.1016/j.cps.2021.03.004. Epub [PubMed PMID: 34051902]
Raposo-Amaral CE, Denadai R, Oliveira YM, Ghizoni E, Raposo-Amaral CA. Apert Syndrome Management: Changing Treatment Algorithm. The Journal of craniofacial surgery. 2020 May/Jun:31(3):648-652. doi: 10.1097/SCS.0000000000006105. Epub [PubMed PMID: 31895846]
Al-Namnam NMN, Hariri F, Rahman ZAA. Distraction osteogenesis in the surgical management of syndromic craniosynostosis: a comprehensive review of published papers. The British journal of oral & maxillofacial surgery. 2018 Jun:56(5):353-366. doi: 10.1016/j.bjoms.2018.03.002. Epub 2018 Apr 13 [PubMed PMID: 29661509]
Hopper RA, Kapadia H, Morton T. Normalizing facial ratios in apert syndrome patients with Le Fort II midface distraction and simultaneous zygomatic repositioning. Plastic and reconstructive surgery. 2013 Jul:132(1):129-140. doi: 10.1097/PRS.0b013e318290fa8a. Epub [PubMed PMID: 23508053]
Mathijssen IM. Guideline for Care of Patients With the Diagnoses of Craniosynostosis: Working Group on Craniosynostosis. The Journal of craniofacial surgery. 2015 Sep:26(6):1735-807. doi: 10.1097/SCS.0000000000002016. Epub [PubMed PMID: 26355968]
Bonfield CM, Shannon CN, Reeder RW, Browd S, Drake J, Hauptman JS, Kulkarni AV, Limbrick DD, McDonald PJ, Naftel R, Pollack IF, Riva-Cambrin J, Rozzelle C, Tamber MS, Whitehead WE, Kestle JRW, Wellons JC, Hydrocephalus Clinical Research Network (HCRN). Hydrocephalus treatment in patients with craniosynostosis: an analysis from the Hydrocephalus Clinical Research Network prospective registry. Neurosurgical focus. 2021 Apr:50(4):E11. doi: 10.3171/2021.1.FOCUS20979. Epub [PubMed PMID: 33794488]
Renier D, Arnaud E, Cinalli G, Sebag G, Zerah M, Marchac D. Prognosis for mental function in Apert's syndrome. Journal of neurosurgery. 1996 Jul:85(1):66-72 [PubMed PMID: 8683284]
Tovetjärn R, Tarnow P, Maltese G, Fischer S, Sahlin PE, Kölby L. Children with Apert syndrome as adults: a follow-up study of 28 Scandinavian patients. Plastic and reconstructive surgery. 2012 Oct:130(4):572e-576e. doi: 10.1097/PRS.0b013e318262f355. Epub [PubMed PMID: 23018718]
Allam KA, Wan DC, Khwanngern K, Kawamoto HK, Tanna N, Perry A, Bradley JP. Treatment of apert syndrome: a long-term follow-up study. Plastic and reconstructive surgery. 2011 Apr:127(4):1601-1611. doi: 10.1097/PRS.0b013e31820a64b6. Epub [PubMed PMID: 21187805]
Khong JJ, Anderson P, Gray TL, Hammerton M, Selva D, David D. Ophthalmic findings in Apert's syndrome after craniofacial surgery: twenty-nine years' experience. Ophthalmology. 2006 Feb:113(2):347-52 [PubMed PMID: 16458095]
Level 2 (mid-level) evidenceCoats DK, Paysse EA, Stager DR. Surgical management of V-pattern strabismus and oblique dysfunction in craniofacial dysostosis. Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and Strabismus. 2000 Dec:4(6):338-42 [PubMed PMID: 11124667]
Level 2 (mid-level) evidenceGoh LC, Azman A, Siti HBK, Khoo WV, Muthukumarasamy PA, Thong MK, Abu Bakar Z, Manuel AM. An audiological evaluation of syndromic and non-syndromic craniosynostosis in pre-school going children. International journal of pediatric otorhinolaryngology. 2018 Jun:109():50-53. doi: 10.1016/j.ijporl.2018.03.010. Epub 2018 Mar 14 [PubMed PMID: 29728184]