Introduction
Hemoglobin is the chief erythrocyte protein that transports oxygen from the lungs to the tissues, carbon dioxide from the tissues to the lungs, and vasodilating nitrous oxide from nitrite to the blood vessels. Hemoglobin consists of α- and β-globins and an iron-bearing tetrapyrrole moiety. α- and β-globins influence oxygen loading and unloading and erythrocyte shape. Abnormal α- and β-globin result in hemoglobin dysfunction, misshapen erythrocytes, erythrocyte fragility, and the systemic symptoms seen in many hemoglobin disorders.
Thalassemia and hemoglobinopathies are collectively the most common Mendelian diseases found in humans.[1] "Thalassemia" refers to quantitative deficiencies of one or more globin subunits, with α-thalassemia and β-thalassemia being defined as reduced or absent production of α-globin and β-globin chains, respectively.[2]
α-thalassemia is a relatively common monogenic blood disorder found worldwide. α-globin production is regulated by 4 α genes located on chromosome 16.[3] α-thalassemia is usually caused by the affected allele's reduced (designated as α+) or complete absence (designated as α°) of globin chain production.[4]
The carrier state can either be an α+ (α-thalassemia 2 resulting from 1 α-globin gene deletion) or α° (α-thalassemia 1 resulting from the deletion of 2 α-globin genes) trait. α-Thalassemia 2 is an asymptomatic carrier state. The 2 other types of α-thalassemia are Hemoglobin H (or HbH, with deletion of 3 α-globin genes) and α-thalassemia major or Hemoglobin Bart's (or Hb Bart's, with deletion of all 4 α-globin genes).[5] Hb Bart's can cause hydrops fetalis.
Thalassemia can be easily confused with iron deficiency anemia (IDA), which needs to be ruled out in a patient with low hemoglobin. Diagnostic tests like complete blood count (CBC) and hemoglobin analysis by high-performance liquid chromatography (HPLC) or electrophoresis may help identify the hemoglobin disorder in patients with chronic anemia (see Image. Hemoglobin Electrophoresis Patterns of Hemoglobin Disorders).[6] More advanced α-thalassemia mutation analytical techniques, such as allele-specific polymerase chain reaction (PCR), reverse dot blot (RDB) analysis, real-time PCR, and DNA sequencing, may also be useful for genetic counseling.[7] Disease management, parental counseling, antenatal diagnosis, newborn screening, and complication prevention are critical for improving the patients' and their families' quality of life.
Types of α-Thalassemia
α-Thalassemia can be classified into 4 types based on the number of functional α-globin genes inherited. The severity of symptoms generally increases with the number of affected genes.
- α-Thalassemia Silent Carrier (αα/α-): arises when 1 α gene is deleted, and α-globin-chain production is enough to ensure a normal hemoglobin synthesis. The patient remains asymptomatic, and the mutation is benign. However, mild anemia can sometimes occur, like increased stress or pregnancy. Patients can pass on the mutation to their progeny.[8]
- α-Thalassemia Minor (αα/–) or (α-/α-): occurs when 2 α genes are deleted. α-globin-chain production is reduced by half. In adults, faster red blood cell (RBC) production can compensate for the reduced α chain production to some extent, thus balancing the amount of α- and β-globin chains. Patients are usually asymptomatic, and anemia is often mild if present.[9]
- HbH Disease (α-/–): results from mating between an α-thalassemia silent carrier and an individual with α-thalassemia minor. HbH disease has 3 α gene deletions, manifesting as anemia of varying degrees. Non-transfusion-dependent (NTDT) or transfusion-dependent (TDT) thalassemia may arise. Excess β-globin chains aggregate to form HbH tetramers. HbH often precipitates within RBCs, damaging RBC membranes and resulting in hemolytic anemia.[10] This disease has a broad phenotypic spectrum and may not be diagnosed until adulthood. Patients may present with splenomegaly, mild jaundice, and characteristic thalassemia-like bone changes due to extramedullary erythropoiesis. Patients may also develop gallstones and experience acute hemolytic episodes following infections or oxidant drug exposure.
- α-Thalassemia Major (–/–): arises from homozygous states and produces the most severe and fatal form of α-thalassemia. This condition is also known as Hb Bart's hydrops fetalis, Hb Bart's, or hydrops fetalis. Excess fetal γ-globin production results from the complete lack of α-globin chains. However, γ-globin forms tetramers with high oxygen affinity and thus does not unload oxygen efficiently in tissues. Generalized edema occurs prenatally, accompanied by severe pleural and pericardial effusions. These manifestations arise from congestive cardiac failure due to severe anemia. Extramedullary erythropoiesis, hepatosplenomegaly, and a large placenta are also seen. The fetus is usually nonviable.[11]
The mother's pregnancy history may provide clues when evaluating patients with possible α-thalassemia.
Etiology and Epidemiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology and Epidemiology
α-globin genes are located on chromosome 16. Humans typically inherit 4 α genes (αα/αα), 2 from each parent. α-thalassemia manifests when α-globin gene deletion in any number occurs. Anemia of varying degrees results from the mutation, as the number of deleted genes affects the amount of α-globin chain produced.[12]
Thalassemia is known to be extremely heterogeneous at the molecular level. Over 200 different mutations are associated with α-thalassemia. The condition is most frequently seen in southeastern and southern Asia, the Middle East, Mediterranean countries, and North and Central Africa. However, increased global movement and mixing of various ethnicities have resulted in the rising thalassemia incidence in Northern Europe and North America.[13] Hb Bart's occurs mainly in Southeast Asia and Southern China.
Pathophysiology
The significant pathophysiological change in thalassemia is imbalanced globin chain production, resulting in RBC fragility. RBC precursors are thus easily destroyed in the bone marrow or peripheral blood. Symptoms include chronic anemia, splenomegaly, and skeletal deformities.[14]
The blood picture is often similar to IDA, with slightly microcytic RBCs. Erythrocytes may be normal in the α-thalassemia silent carrier type. α-thalassemia 1 usually presents with mild anemia, slight RBC index decreases (mean corpuscular volume or MCV and mean corpuscular hemoglobin or MCH), hypochromia, microcytosis, and anisopoikilocytosis. HbA2 level is in the low to low-normal range (1.5%-2.5%).
During the neonatal period, moderate amounts of Hb Bart's (3% to 8%) can be seen on blood films. A few (1:1,000-10,000 RBCs) intracellular hemoglobin precipitates forming Heinz inclusion bodies may also be detected. Hematological parameters should be reevaluated after iron supplementation.
HbH disease generally occurs as NTDT, with α-globin synthesis reduced by about a quarter of the expected level. The presence of HbH, which contains β-globin chain homotetramers, can be detected by HPLC or electrophoresis.[15] HbH amount is between 3% and 30% and is associated with mild-to-severe microcytic or normocytic anemia. Elevated bilirubin levels arise due to a moderate hemolytic component.[16]
The most severe α-thalassemia form is the homozygous α°-thalassemia state or Hb Bart's hydrops fetalis syndrome. The fetus cannot synthesize any α-globin chains to make HbF or HbA. Fetal blood shows the presence of only Hb Bart's (γ4) and some amount of embryonic Hemoglobin Portland (Hb Portland). Prenatal diagnosis is crucial for parental counseling in such cases.[17]
TDT patients develop various complications due to systemic iron overload. Hemoglobin denaturation causes iron deposition in RBC membranes, resulting in membrane weakening and hemolysis. The concomitant effects of ineffective erythropoiesis, chronic anemia, and hypoxia increase gastrointestinal iron absorption. All these factors also cause increased iron deposition in tissues, resulting in hemosiderosis. Free iron generates reactive oxygen species, leading to free radical-mediated tissue damage, organ dysfunction, and subsequent organ failure. TDT patients require regular iron chelation therapy and iron level monitoring to avoid such complications.[18]
Specimen Requirements and Procedure
Specimen collection depends on the choice of diagnostic test. α-thalassemia type identification requires a battery of various laboratory tests, including CBC. Hemoglobin analysis identifies and quantifies various hemoglobin types, including HbH, Hb Bart's, and non-gene deletion mutants associated with more severe symptoms like Hb Constant Spring (HbCS). EDTA vials are used for whole-blood collection for CBC, hemoglobin analysis, and molecular testing.[19]
Cytogenetic analysis on chorionic villi samples or amniotic fluid cells is the procedure of choice for prenatal α-thalassemia diagnosis in suspected carriers or thalassemia minor parents. The intact chorionic villi can be preserved in a culture medium for up to 7 days.[20][21]
Diagnostic Tests
The initial diagnostic tests for α-thalassemia should include the following:
- CBC: This lab testing may be done using automated laboratory hematology analyzers. Differentiating the thalassemia trait from IDA or α- from β-thalassemia cannot be done by only determining the hemoglobin level, MCV, and MCH concentration (MCHC). However, RBC count elevation can distinguish α-thalassemia from IDA, which often has a low RBC count.[22] Thalassemia does not directly affect the platelet and WBC counts.
- Iron studies: Serum ferritin and transferrin levels can differentiate thalassemia from IDA. A normal or slightly increased ferritin level with nearly normal transferrin is more consistent with thalassemia than IDA, which often manifests with low ferritin and elevated transferrin.
- Peripheral smear: This lab result shows microcytic, hypochromic anemia, target cells, teardrop cells, and cells with basophilic stippling in thalassemia cases, though these findings are also common in IDA. The presence of golf-ball-like hemoglobin inclusions indicates HbH disease.
- Hemoglobin analysis: HPLC or electrophoresis can quantify the different hemoglobin types in the patient’s blood (see Image. α-Thalassemia Major on Hemoglobin Electrophoresis). These techniques can also help differentiate different thalassemia types with good precision and reproducibility.[23]
- Molecular testing: allele-specific PCR, RDB analysis, gap-PCR, real-time PCR with melting curve analysis, and DNA sequencing can help identify the mutation causing α-thalassemia. Molecular testing is essential to genetic counseling and therapeutic research.[24][25]
Test results should be interpreted in conjunction with clinical findings to guide treatment decisions.
Testing Procedures
High-throughput techniques offer the advantages of speed, efficiency, and scalability, making them valuable tools in both research and clinical settings for detecting and characterizing α-thalassemia mutations. The choice of technique depends on factors such as the desired resolution level and available resources.
High-Performance Liquid Chromatography
HPLC separates, identifies, and quantifies different hemoglobin molecules. The specimen is passed through a slightly negatively charged silica gel column. Highly positively charged hemoglobin variants have more affinity for the column and thus elute after less positively charged hemoglobin types. Inorganic phosphate buffers help during elution.[26] The chromatograms generated by HPLC show the retention time, peak, and percentage of different hemoglobin fractions, allowing for the accurate diagnosis of hemoglobinopathies and thalassemia.[27]
Using inorganic phosphate buffers in the HPLC system is essential for maintaining mobile phase pH and effective separation of hemoglobin variants. The buffer and its pH must be carefully selected to ensure optimal chromatographic conditions and prevent damage to the column hardware and bed.[28]
Capillary Electrophoresis System
Hemoglobin components are separated using silica capillaries in an alkaline buffer medium. Hemoglobin is negatively charged in alkaline solutions and thus migrates to the anode. The structural hemoglobin variants, having different surface charges, separate. The photometry technique then determines the amounts of each variant at a wavelength of 415 nm. Capillary electrophoresis can be used for both prenatal and postnatal hemoglobinopathy diagnosis.[29] Both HPLC and capillary electrophoresis can detect HbH, Hb Bart’s, and HbCS.[30]
Allele-Specific Polymerase Chain Reaction
PCR utilizes 2 primers with identical sequences except at the 3′-terminus base. One of the primer 3’ bases is complementary to the wild-type, and the other complements the mutant base. A common primer for the opposite strand is also used. Taq polymerase amplifies the genes but needs perfect matching of the primer 3′-end with the DNA template.
In healthy individuals, the PCR product is observed only in the reaction where the wild-type primer set is present. In contrast, a heterozygote produces a band in both mutant and wild-type primer sets. Individuals with homozygous mutations are negative in the wild-type and positive in the mutant primer set.[31]
Reverse Dot Blot Analysis
The PCR product may be transferred on a membrane filter sheet as a dot to identify a suspected mutation more easily. This PCR product can then hybridize with an allele-specific oligomeric DNA probe. The probe can be radiolabeled with phosphorus for autoradiography or other reporter groups like biotin. Alternatively, an enzyme such as horseradish peroxidase may be attached. The product may be visualized using a chemiluminescent or colorimetric reaction.
A healthy individual normally shows positive dots with each wild-type probe but not with any of the mutant probes. Meanwhile, heterozygotes often have a mixed pattern, demonstrating hybridization with both mutant and normal dots. Homozygotes typically demonstrate hybridization with the mutant probe only and not with normal probes.
Real-Time PCR with Melting Curve Analysis
Real-time PCR is often less time-consuming and labor-intensive than conventional PCRs. Real-time PCR is widely used to detect, characterize, and quantify nucleic acids. This technology also has a low risk of post-PCR contamination. Real-time PCR for thalassemia diagnosis uses 2 approaches: intercalating dye and probe-based assays. A fluorescent signal is obtained from PCR synthesis of the product. Dyes like SYBR® Green are used. The melting curve analysis is useful in distinguishing α-thalassemia 1 and α-thalassemia 2 heterozygotes, HbH disease, and α-thalassemia 1 homozygotes (Hb Bart's).[32]
Direct DNA Sequencing
PCR product sequencing can help in identifying the specific gene mutations in α-thalassemia. Sanger’s dideoxy termination method is the most widely used.[33]
Multiplex Ligation-Dependent Probe Amplification
This method uses multiplex PCR to detect duplications or deletions in the screened region. One advantage of this technique is that it can find known and unknown deletions in unsolved cases when conventional techniques fail. Multiplex ligation-dependent probe amplification is easy to use, and only a thermocycler and electrophoresis equipment are needed.[34][35]
Next-Generation Sequencing
Next-generation sequencing (NGS) technology can sequence the entire human genome in an ultra-high throughput manner at high speed. The target NGS approach may be used to analyze entire globin gene-coding regions, key regulatory regions, and modifier genes.[36] NGS may be more accurate than conventional thalassemia diagnostic methods like CBC and hemoglobin analysis and typing. This technology increases gene sequencing capacity from a few hundred to several thousand base pairs in a single analysis. The additional advantages of NGS include lower sample input requirements, higher accuracy, and the ability to detect variants at lower allele frequencies than Sanger sequencing.[37]
Interfering Factors
Elements that can compromise accuracy can reduce the utility of the above diagnostic tests. Sources of errors are broadly categorized as preanalytical, analytical, and postanalytical. Preanalytical factors consist of lapses following the standard protocols during collecting, transporting, or storing specimens like blood or chorionic villi samples. Such lapses can introduce errors in the method, rendering the results inaccurate. For example, hemoglobin can denature if stored for long periods or at high temperatures. Poor sample preparation, as in failing to remove maternal tissue from chorionic villi specimens, can also produce measurement errors.
During analysis, using improperly stored or prepared buffers, reagents, or stains can contribute to inaccurate analysis. Rounding off reported figures is an example of a postanalytical factor that can introduce diagnostic errors.[38]
The accessibility of these diagnostic tests is another factor that can reduce their utility. Techniques like NGS, allele-specific PCR, and multiplex ligation-dependent probe amplification are not feasible for widespread use in diverse populations due to their high cost and laborious maintenance.
Results, Reporting, and Critical Findings
Healthy adults usually have the maximum amount of hemoglobin A (HbA), comprising 95% to 98% of their total blood hemoglobin. Variants like hemoglobin A2 (HbA2) constitute 2% to 3%, while hemoglobin F (HbF) consists of less than 2% of total adult hemoglobin. All α-thalassemia-affected individuals show varying degrees of anemia that may manifest as low hemoglobin, MCH, and MCV and normal to slightly decreased HbA2 levels.
α-Thalassemia Carriers
α trait is an asymptomatic carrier state. RBC levels may be normal, though with slight microcytosis. A minor amount (1% to 3%) of Hb Bart's may sometimes be detected in the neonatal period.
α° trait is usually marked by slight anemia, slightly reduced MCV and MCH, RBC microcytosis, hypochromia, and anisopoikilocytosis. Hb Bart's may be detected in moderate amounts (3% to 8%) during the neonatal period. Some Heinz inclusion bodies may also be noted.
α-thalassemia carriers often have increased RBC count, which helps differentiate it from IDA (see Image. Thalassemia and Iron-Deficiency Anemia Laboratory Differentiation). Clinically asymptomatic cases are usually diagnosed during a routine health checkup or antenatal screening.
α-Thalassemia Minor
α-Thalassemia minor results from 2 α-chain gene deletions. Patients have mild-to-moderate anemia, with hemoglobin levels usually ranging from 7 to 10 g/dL. HbA2 levels range from 3% to 3.5%, while HbF levels range between 10% and 50%. Molecular testing is a more accurate diagnostic tool.[39]
α-Thalassemia Intermedia (HbH Disease)
This condition's predominant characteristic is anemia with varying HbH amounts (0.8% to 40%). The mutation type, whether deletional or nondeletional, affects clinical severity. Patients often present with severe microcytic or normocytic anemia early in life. Hemoglobin levels below 7 g/dL may be reported. MCH is also often low at less than 20 pg, and the peripheral blood film typically shows poikilocytosis with tear-drop cells, increased erythroblasts, and target cells.[19]
In adults, the low α-globin chain levels cause excess β-globin chains to form the β tetramers comprising HbH (0.8% to 40%), detected as inclusion bodies on microscopy. These inclusion bodies may be identified by 1% brilliant cresyl blue staining. Routine hemoglobin analysis can also detect HbH if present in large quantities.[40]
Meanwhile, the laboratory findings in HbH deletional types demonstrate microcytic, hypochromic anemia and inclusion bodies.[19] HbH and Hb Bart's appear as fast-moving hemoglobin on electrophoresis or HPLC in these patients. Nondeletional HbH types like HbCS have more severe presentations, as erythropoiesis is largely ineffective. Hemoglobin is often severely low, about 2 g/dL on average.[40]
α-Thalassemia Major (Hb Bart's Hydrops Fetalis Syndrome)
Hb Bart's hydrops fetalis syndrome is notable for the presence of Hb Bart's and the absence of HbF. α-globin synthesis lower than 70% of the normal in the fetal period induces tetramerization of excess γ-globin chains to form Hb Bart's, which may be identified on routine Hb analysis. Nonfunctional γ and β homotetramers constitute most of the hemoglobin in the fetal erythrocytes. The only functional hemoglobin in these infants is Hb Portland (ζγ), which acts as the oxygen carrier.[40]
Mothers pregnant with a fetus with Hb Bart's hydrops fetalis usually present clinically between 20 and 26 weeks of gestation with pregnancy-induced hypertension and polyhydramnios. Fetal ultrasound should reveal hydrops. On Doppler measurement, fetal anemia is suggested by an increase in the middle cerebral artery's peak systolic velocity compared to the gestation-specific reference interval. Fetal blood sampling by cordocentesis typically demonstrates severe fetal anemia, with hemoglobin less than 80 g/L.[41] Other possible causes of hydrops fetalis must also be ruled out, such as toxoplasmosis, rubella, cytomegalovirus, herpes simplex, parvovirus B19 infection, and hemolytic disease of the fetus due to red cell alloantibodies.[42]
HPLC of a cordocentesis blood sample shows 1 or 2 very sharp and narrow peaks at the chromatogram injection point.[43] The major band is Hb Bart's, while the smaller band is attributable to Hb Portland. The complete absence of HbF should be noted. Alkaline electrophoresis shows the Hb Bart's band migrating to the anodal position and the Hb Portland band moving to the HbA position.[11]
Hb Bart's hydrops fetalis is almost invariably fatal if left untreated, with some fetuses dying in utero and others surviving a few hours after birth. Treatment with intrauterine transfusion may be able to salvage the fetus. However, the risk of complications like growth retardation and severe brain damage remains, which may be related to hypoxemia following long-standing intrauterine anemia.[41]
Laboratory investigation of the parents of fetuses with Hb Bart's hydrops fetalis shows a typical HPLC pattern with normal HbF and HbA2 quantification.[44] Parental analysis typically shows a decreased hemoglobin concentration, MCH, and MCV. The blood smear also typically shows hypochromic, microcytic red cells (see Image. Peripheral Blood Smear for a Case of Hydrops Fetalis). The HbH inclusion body test is usually positive in the parents.[43]
Clinical Significance
α-thalassemia has a broad spectrum of clinical presentations. Accurate diagnosis is thus crucial to management decision-making, as in determining whether a fetus with Hb Bart's should be aborted or given intrauterine transfusions.[45]
Parents with the silent carrier or α-thalassemia minor type may be properly counseled if diagnosed. Techniques like molecular analysis may be used in clinically difficult cases, as in families with mild mutations or α and β interactions. The laboratory evaluation of α-thalassemia thus holds paramount importance in improving patients' quality of life by timely initiation of therapy, counseling sessions, blood transfusions if required, and prevention of future complications.
Quality Control and Lab Safety
Laboratory analytical quality factors should be guided by a facility's definition and criteria for quality. Several errors can arise from external and internal sources and permanent and variable factors.[46] Establishing internal quality control and external quality assurance systems (EQAS) is imperative for laboratories to uphold testing methodology precision, reliability, and quality standards.[47] EQAS serves as a crucial mechanism for evaluating a laboratory's performance and fostering continuous improvement through external assessment and comparison with other laboratories.[48]
In-house standard operating procedures must be established, to which laboratory personnel should strictly comply. Even a slight change in quality parameters can make a significant difference and render erroneous results.[49] Reference standards and control materials must be meticulously noted during analysis. Method-specific controls and standards may be prepared in-house or obtained commercially. Laboratories should establish a reference range for all specific parameters analyzed, but such references should not be significantly different from published data.[50]
Monitoring is required at each step to ensure compliance with established protocols and check for deviations from the normal. The Levey Jennings chart may aid in the monitoring process. This chart is a graphic representation of daily controls or averages of the results. Thus, the method can help identify and correct single-day errors.[51]
Every clinical laboratory should implement a formal safety program as its top priority to ensure patient and laboratory staff safety. Such a program should be the foundation for recognizing, reducing, and controlling potential laboratory hazards. Adhering to strict safety protocols significantly diminishes the chances of errors, incidents, and exposure to unsafe materials.[52]
Protective gear such as gloves, masks, protective eyewear, and gowns must be employed when collecting patient specimens. Disposable gloves made from nonsterile latex or alternative materials offering adequate barrier protection should be used. Phlebotomists should change gloves when moving between blood draws from different patients. Hand hygiene protocols are essential when changing gloves.[53]
Facial protection is necessary for lab technicians when the risk of blood or bodily fluid contamination is considerable. Minimizing syringe use and disposing of needles in rigid containers is advised. Wearing protective clothing is recommended to safeguard against potentially infectious substances. Removing protective gear is essential before leaving the laboratory.[54]
Enhancing Healthcare Team Outcomes
α-thalassemia diagnosis and management involve a collaborative effort from an interprofessional healthcare team. Members of the team may include the following:
- Hematologist: responsible for diagnosing and managing α-thalassemia, interpreting laboratory results, and overseeing treatment plans.
- Genetic counselor: provides information and counseling to individuals and families about the genetic aspects of α-thalassemia. This professional also helps assess the risk of transmitting the condition to the offspring, explains genetic test results, and assists with family planning decisions.
- Laboratory technologist: conducts various laboratory tests, including CBC, hemoglobin electrophoresis, molecular genetic testing, and other specialized assays, ensuring analytical accuracy and report timeliness.
- Pediatrician or internal medicine physician: involved in the overall care and management of individuals with α-thalassemia.
- Obstetrician/gynecologist: monitors high-risk pregnancies and addresses potential complications, such as fetal hydrops.
- Nurse practitioner: helps provide ongoing care, education, and support to individuals with α-thalassemia and their families.
- Pharmacist: collaborates with the healthcare team to manage medications, such as iron chelators or other supportive therapies.
- Transfusion specialist: may be involved in managing blood transfusions for individuals with more severe forms of α-thalassemia.
- Clinical geneticist: consulted for complex genetic cases or when additional expertise is required to interpret genetic testing results.
Effective communication and collaboration among these professionals are crucial for providing comprehensive and coordinated care for individuals with α-thalassemia. A holistic, patient-centered approach helps ensure the best possible patient outcomes.
Media
(Click Image to Enlarge)
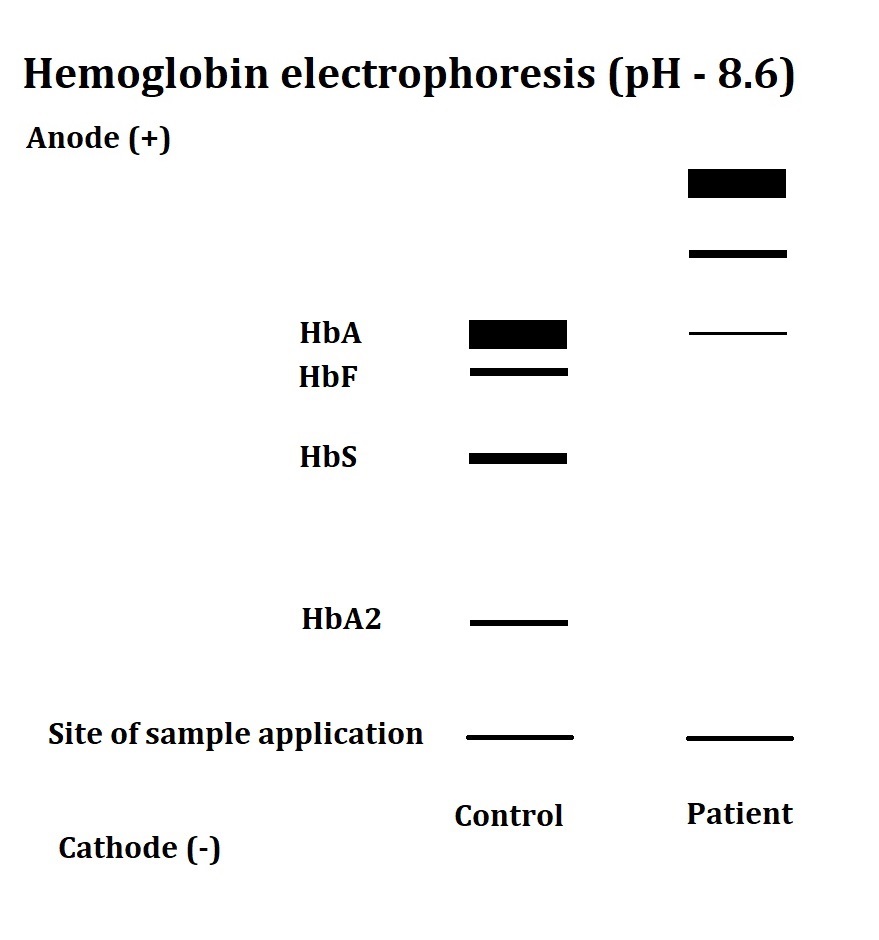
α-Thalassemia Major on Hemoglobin Electrophoresis. This hemoglobin electrophoresis result shows the presence of HbH and Hb Bart's with scanty HbA in the patient specimen.
Contributed by Amit Sonagra, MD
(Click Image to Enlarge)
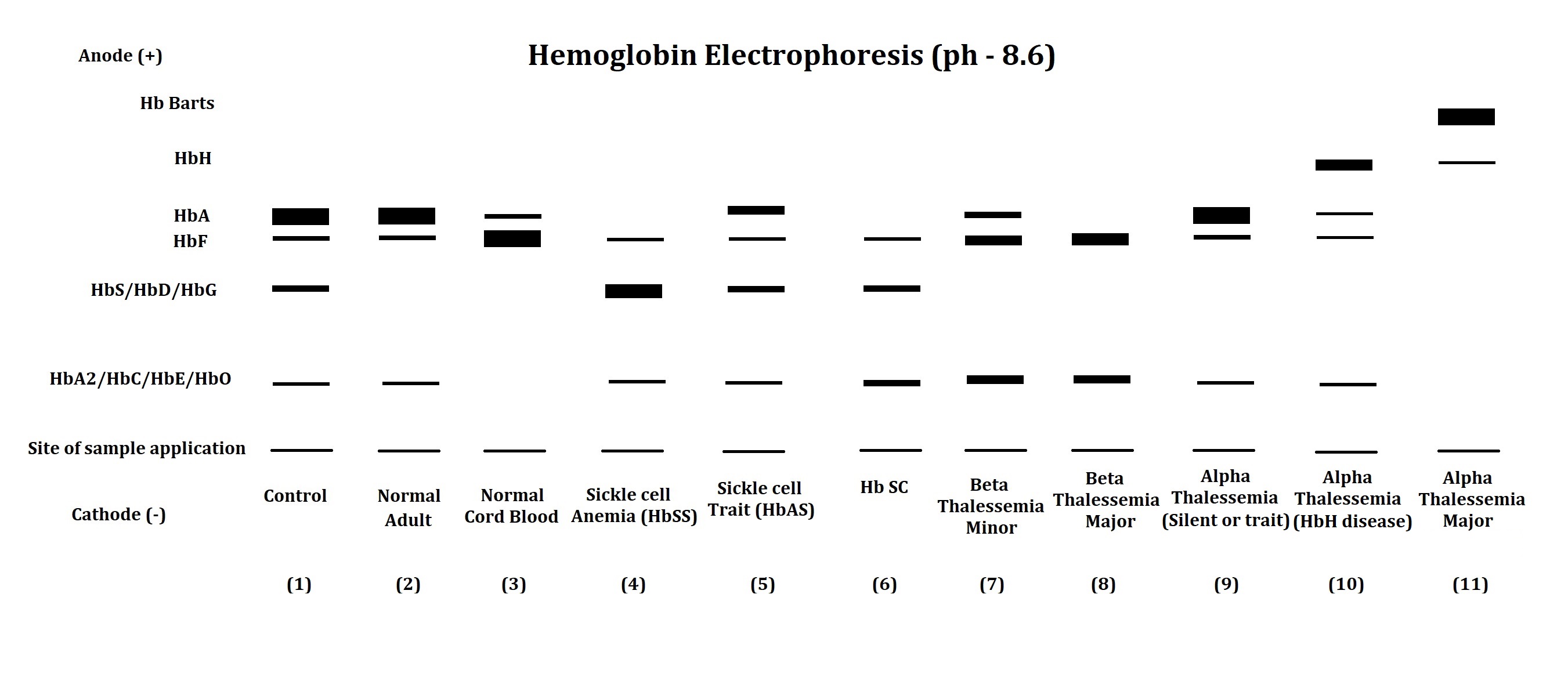
Hemoglobin Electrophoresis Patterns of Hemoglobin Disorders. This image shows the typical hemoglobin electrophoresis results for various thalassemia types and hemoglobinopathies.
Contributed by Amit Sonagra, MD
(Click Image to Enlarge)
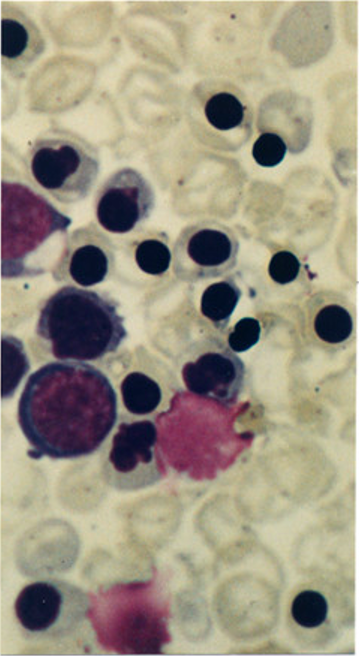
Peripheral Blood Smear for a Case of Hydrops Fetalis. This image shows a peripheral blood film with immature red-cell precursors and red blood cells with hypochromia, microcytosis, anisocytosis, and poikilocytosis. Clinical findings are consistent with Hemoglobin Bart's hydrops fetalis syndrome.
Used with Permission from BMC Publishing
(Click Image to Enlarge)
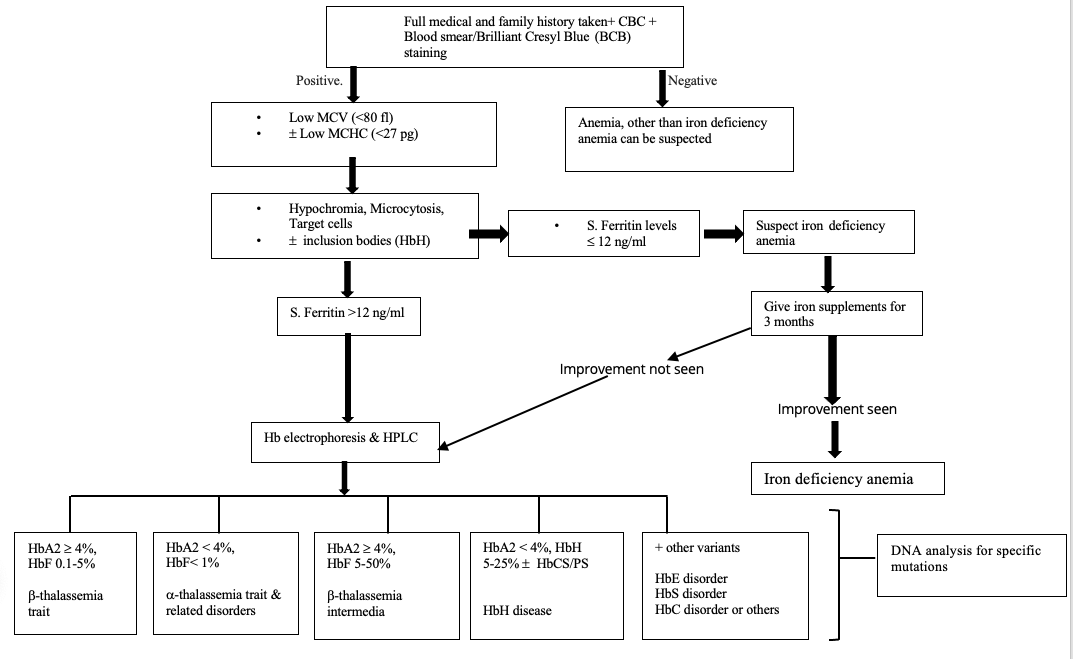
Thalassemia and Iron-Deficiency Anemia Laboratory Differentiation. This image shows a flow chart for the laboratory evaluation and diagnosis of thalassemia and its differentiation from iron-deficiency anemia.
Contributed by Anita Motiani, MD
References
Thein SL. Molecular basis of β thalassemia and potential therapeutic targets. Blood cells, molecules & diseases. 2018 May:70():54-65. doi: 10.1016/j.bcmd.2017.06.001. Epub 2017 Jun 20 [PubMed PMID: 28651846]
Thein SL. The molecular basis of β-thalassemia. Cold Spring Harbor perspectives in medicine. 2013 May 1:3(5):a011700. doi: 10.1101/cshperspect.a011700. Epub 2013 May 1 [PubMed PMID: 23637309]
Level 3 (low-level) evidenceBernini LF, Harteveld CL. Alpha-thalassaemia. Bailliere's clinical haematology. 1998 Mar:11(1):53-90 [PubMed PMID: 10872473]
Harewood J, Azevedo AM. Alpha Thalassemia. StatPearls. 2024 Jan:(): [PubMed PMID: 28722856]
Weatherall DJ. The Evolving Spectrum of the Epidemiology of Thalassemia. Hematology/oncology clinics of North America. 2018 Apr:32(2):165-175. doi: 10.1016/j.hoc.2017.11.008. Epub [PubMed PMID: 29458724]
Munkongdee T, Chen P, Winichagoon P, Fucharoen S, Paiboonsukwong K. Update in Laboratory Diagnosis of Thalassemia. Frontiers in molecular biosciences. 2020:7():74. doi: 10.3389/fmolb.2020.00074. Epub 2020 May 27 [PubMed PMID: 32671092]
Rund D. Thalassemia 2016: Modern medicine battles an ancient disease. American journal of hematology. 2016 Jan:91(1):15-21. doi: 10.1002/ajh.24231. Epub [PubMed PMID: 26537527]
Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassemias. Haematologica. 2013 Jun:98(6):833-44. doi: 10.3324/haematol.2012.066845. Epub [PubMed PMID: 23729725]
Piel FB, Weatherall DJ. The α-thalassemias. The New England journal of medicine. 2014 Nov 13:371(20):1908-16. doi: 10.1056/NEJMra1404415. Epub [PubMed PMID: 25390741]
Poyart C, Wajcman H. Hemolytic anemias due to hemoglobinopathies. Molecular aspects of medicine. 1996 Apr:17(2):129-42 [PubMed PMID: 8813715]
He S, Li J, Huang P, Zhang S, Lin L, Zuo Y, Tian X, Zheng C, Qiu X, Chen B. Characterization of Hb Bart's Hydrops Fetalis Caused by - -(SEA) and a Large Novel α(0)-Thalassemia Deletion. Hemoglobin. 2018 Jan:42(1):61-64. doi: 10.1080/03630269.2018.1434198. Epub 2018 Mar 1 [PubMed PMID: 29493331]
Level 2 (mid-level) evidencePang W, Long J, Weng X, Fan Q, Sun L, Pan Z, Fan Z. Identification of Three Types of α-Thalassemia Deletion, -α(21.9), -α(2.4), and - -(THAI), and Their Frequencies, in One Family in the Population of Southern Guangxi Zhuang Autonomous Region, People's Republic of China. Hemoglobin. 2018 Jan:42(1):37-42. doi: 10.1080/03630269.2018.1428618. Epub 2018 Feb 15 [PubMed PMID: 29447013]
Williams TN, Weatherall DJ. World distribution, population genetics, and health burden of the hemoglobinopathies. Cold Spring Harbor perspectives in medicine. 2012 Sep 1:2(9):a011692. doi: 10.1101/cshperspect.a011692. Epub 2012 Sep 1 [PubMed PMID: 22951448]
Level 3 (low-level) evidenceKalfa TA. Diagnosis and clinical management of red cell membrane disorders. Hematology. American Society of Hematology. Education Program. 2021 Dec 10:2021(1):331-340. doi: 10.1182/hematology.2021000265. Epub [PubMed PMID: 34889366]
Bhat VS, Dewan KK, Krishnaswamy PR. The Diagnosis of α-Thalassaemia: A Case of Hemoglobin H -α Deletion. Indian journal of clinical biochemistry : IJCB. 2010 Oct:25(4):435-40. doi: 10.1007/s12291-010-0053-7. Epub 2010 Sep 14 [PubMed PMID: 21966120]
Level 3 (low-level) evidenceViprakasit V, Limwongse C, Sukpanichnant S, Ruangvutilert P, Kanjanakorn C, Glomglao W, Sirikong M, Utto W, Tanphaichitr VS. Problems in determining thalassemia carrier status in a program for prevention and control of severe thalassemia syndromes: a lesson from Thailand. Clinical chemistry and laboratory medicine. 2013 Aug:51(8):1605-14. doi: 10.1515/cclm-2013-0098. Epub [PubMed PMID: 23525874]
King AJ, Higgs DR. Potential new approaches to the management of the Hb Bart's hydrops fetalis syndrome: the most severe form of α-thalassemia. Hematology. American Society of Hematology. Education Program. 2018 Nov 30:2018(1):353-360. doi: 10.1182/asheducation-2018.1.353. Epub [PubMed PMID: 30504332]
Giordano PC. Strategies for basic laboratory diagnostics of the hemoglobinopathies in multi-ethnic societies: interpretation of results and pitfalls. International journal of laboratory hematology. 2013 Oct:35(5):465-79. doi: 10.1111/ijlh.12037. Epub 2012 Dec 7 [PubMed PMID: 23217050]
Brancaleoni V, Di Pierro E, Motta I, Cappellini MD. Laboratory diagnosis of thalassemia. International journal of laboratory hematology. 2016 May:38 Suppl 1():32-40. doi: 10.1111/ijlh.12527. Epub 2016 May 16 [PubMed PMID: 27183541]
Hansson K, Schuring-Blom GH, Leschot NJ. The preserving of chorionic villi before establishing long-term cell cultures for cytogenetic analysis. Prenatal diagnosis. 1995 Nov:15(11):1067-9 [PubMed PMID: 8606886]
Old J, Henderson S. Molecular diagnostics for haemoglobinopathies. Expert opinion on medical diagnostics. 2010 May:4(3):225-40. doi: 10.1517/17530051003709729. Epub 2010 Mar 15 [PubMed PMID: 23488532]
Level 3 (low-level) evidenceGalanello R, Origa R. Beta-thalassemia. Orphanet journal of rare diseases. 2010 May 21:5():11. doi: 10.1186/1750-1172-5-11. Epub 2010 May 21 [PubMed PMID: 20492708]
Oyaert M, Van Laer C, Claerhout H, Vermeersch P, Desmet K, Pauwels S, Kieffer D. Evaluation of the Sebia Minicap Flex Piercing capillary electrophoresis for hemoglobinopathy testing. International journal of laboratory hematology. 2015 Jun:37(3):420-5. doi: 10.1111/ijlh.12305. Epub 2014 Oct 16 [PubMed PMID: 25324031]
Chong SS, Boehm CD, Higgs DR, Cutting GR. Single-tube multiplex-PCR screen for common deletional determinants of alpha-thalassemia. Blood. 2000 Jan 1:95(1):360-2 [PubMed PMID: 10607725]
Liu YT, Old JM, Miles K, Fisher CA, Weatherall DJ, Clegg JB. Rapid detection of alpha-thalassaemia deletions and alpha-globin gene triplication by multiplex polymerase chain reactions. British journal of haematology. 2000 Feb:108(2):295-9 [PubMed PMID: 10691858]
Rao S, Kar R, Gupta SK, Chopra A, Saxena R. Spectrum of haemoglobinopathies diagnosed by cation exchange-HPLC & modulating effects of nutritional deficiency anaemias from north India. The Indian journal of medical research. 2010 Nov:132(5):513-9 [PubMed PMID: 21150000]
Kunwandee J, Srivorakun H, Fucharoen G, Sanchaisuriya K, Fucharoen S. ARKRAY ADAMS A1c HA-8180T Analyzer for Diagnosis of Thalassemia and Hemoglobinopathies Common in Southeast Asia. Laboratory medicine. 2014 Summer:45(3):e112-21. doi: 10.1309/LMMH649POETQREXL. Epub [PubMed PMID: 25217515]
Khera R, Singh T, Khuana N, Gupta N, Dubey AP. HPLC in characterization of hemoglobin profile in thalassemia syndromes and hemoglobinopathies: a clinicohematological correlation. Indian journal of hematology & blood transfusion : an official journal of Indian Society of Hematology and Blood Transfusion. 2015 Mar:31(1):110-5. doi: 10.1007/s12288-014-0409-x. Epub 2014 Jun 5 [PubMed PMID: 25548455]
Borbely N, Phelan L, Szydlo R, Bain B. Capillary zone electrophoresis for haemoglobinopathy diagnosis. Journal of clinical pathology. 2013 Jan:66(1):29-39. doi: 10.1136/jclinpath-2012-200946. Epub 2012 Oct 26 [PubMed PMID: 23105123]
Khongthai K, Ruengdit C, Panyasai S, Pornprasert S. Analysis of Deletional Hb H Diseases in Samples with Hb A(2)-Hb H and Hb A(2)-Hb Bart's on Capillary Electrophoresis. Hemoglobin. 2019 Jul-Sep:43(4-5):245-248. doi: 10.1080/03630269.2019.1683573. Epub 2019 Nov 5 [PubMed PMID: 31687860]
Suwannakhon N, Pangeson T, Seeratanachot T, Mahingsa K, Pingyod A, Bumrungpakdee W, Sanguansermsri T. Noninvasive prenatal screening test for compound heterozygous beta thalassemia using an amplification refractory mutation system real-time polymerase chain reaction technique. Hematology reports. 2019 Sep 18:11(3):8124. doi: 10.4081/hr.2019.8124. Epub 2019 Sep 18 [PubMed PMID: 31579144]
Qin J, He J, Li Y, Liu N, Tao F, Zhang P, Guo W, Qin Q, Zhou W. One-step genotyping of α-thalassaemia by multiplex symmetric PCR melting curve. Journal of clinical pathology. 2023 Sep:76(9):632-636. doi: 10.1136/jclinpath-2022-208363. Epub 2022 Jun 14 [PubMed PMID: 35701141]
Level 2 (mid-level) evidenceGreen RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O'Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG, American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2013 Jul:15(7):565-74. doi: 10.1038/gim.2013.73. Epub 2013 Jun 20 [PubMed PMID: 23788249]
Level 1 (high-level) evidenceZhou YQ, Xiao GF, Li LY, Li WD, Liu ZY, Zhu LF, Mo QH, Qu XJ, Xu XM. Evaluation of clinical application of gap-PCR as a routine method for alpha-thalassemia carrier detection. Di 1 jun yi da xue xue bao = Academic journal of the first medical college of PLA. 2002 May:22(5):434-6 [PubMed PMID: 12390707]
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic acids research. 2002 Jun 15:30(12):e57 [PubMed PMID: 12060695]
Slatko BE, Gardner AF, Ausubel FM. Overview of Next-Generation Sequencing Technologies. Current protocols in molecular biology. 2018 Apr:122(1):e59. doi: 10.1002/cpmb.59. Epub [PubMed PMID: 29851291]
Level 3 (low-level) evidenceEmms A, Castleman J, Allen S, Williams D, Kinning E, Kilby M. Next Generation Sequencing after Invasive Prenatal Testing in Fetuses with Congenital Malformations: Prenatal or Neonatal Investigation. Genes. 2022 Aug 24:13(9):. doi: 10.3390/genes13091517. Epub 2022 Aug 24 [PubMed PMID: 36140685]
Kingchaiyaphum B, Sanchaisuriya K, Fucharoen G, Chaibunruang A, Hess SY, Hinnouho GM, Barffour MA, Wessells KR, Kounnavong S, Fucharoen S. Hemoglobins F, A(2) , and E levels in Laotian children aged 6-23 months with Hb E disorders: Effect of age, sex, and thalassemia types. International journal of laboratory hematology. 2020 Jun:42(3):277-283. doi: 10.1111/ijlh.13164. Epub 2020 Feb 12 [PubMed PMID: 32048804]
Todd D, Lai MC, Beaven GH, Huehns ER. The abnormal haemoglobins in homozygous alpha-thalassaemia. British journal of haematology. 1970 Jul:19(1):27-31 [PubMed PMID: 5465779]
Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet journal of rare diseases. 2010 May 28:5():13. doi: 10.1186/1750-1172-5-13. Epub 2010 May 28 [PubMed PMID: 20507641]
Curran M, Mikhael M, Sun WD, Lim J, Leung A, Morchi G, Chmait RH. Perinatal Management of Bart's Hemoglobinopathy: Paradoxical Effects of Intrauterine, Transplacental, and Partial Exchange Transfusions. AJP reports. 2020 Jan:10(1):e11-e14. doi: 10.1055/s-0039-3401799. Epub 2020 Jan 27 [PubMed PMID: 31993246]
Songdej D, Babbs C, Higgs DR, BHFS International Consortium. An international registry of survivors with Hb Bart's hydrops fetalis syndrome. Blood. 2017 Mar 9:129(10):1251-1259. doi: 10.1182/blood-2016-08-697110. Epub 2017 Jan 5 [PubMed PMID: 28057638]
Ghosh K, Colah R, Manglani M, Choudhry VP, Verma I, Madan N, Saxena R, Jain D, Marwaha N, Das R, Mohanty D, Choudhary R, Agarwal S, Ghosh M, Ross C. Guidelines for screening, diagnosis and management of hemoglobinopathies. Indian journal of human genetics. 2014 Apr:20(2):101-19. doi: 10.4103/0971-6866.142841. Epub [PubMed PMID: 25400338]
Torcharus K, Sriphaisal T, Krutvecho T, Ketupanya A, Vuthiwong C, Suwanasophon C, Noonai A. Prenatal diagnosis of Hb Bart's hydrops fetalis by PCR technique: Pramongkutklao experience. The Southeast Asian journal of tropical medicine and public health. 1995:26 Suppl 1():287-90 [PubMed PMID: 8629126]
Dame C, Albers N, Hasan C, Bode U, Eigel A, Hansmann M, Brenner R, Bartmann P. Homozygous alpha-thalassaemia and hypospadias--common aetiology or incidental association? Long-term survival of Hb Bart's hydrops syndrome leads to new aspects for counselling of alpha-thalassaemic traits. European journal of pediatrics. 1999 Mar:158(3):217-20 [PubMed PMID: 10094442]
Level 3 (low-level) evidenceFenta DA, Ali MM. Factors Affecting Quality of Laboratory Result During Ordering, Handling, and Testing of the Patient's Specimen at Hawassa University College of Medicine and Health Science Comprehensive Specialized Hospital. Journal of multidisciplinary healthcare. 2020:13():809-821. doi: 10.2147/JMDH.S264671. Epub 2020 Aug 18 [PubMed PMID: 32922023]
Level 2 (mid-level) evidenceKinns H, Pitkin S, Housley D, Freedman DB. Internal quality control: best practice. Journal of clinical pathology. 2013 Dec:66(12):1027-32. doi: 10.1136/jclinpath-2013-201661. Epub 2013 Sep 26 [PubMed PMID: 24072731]
Level 2 (mid-level) evidenceLaudus N, Nijs L, Nauwelaers I, Dequeker EMC. The Significance of External Quality Assessment Schemes for Molecular Testing in Clinical Laboratories. Cancers. 2022 Jul 28:14(15):. doi: 10.3390/cancers14153686. Epub 2022 Jul 28 [PubMed PMID: 35954349]
Level 2 (mid-level) evidenceSkitek M, Martinello F, Jerin A. How to Really Understand and Improve the System of Internal Quality Control and External Quality Assessment in the Accreditation Process of the Medical Laboratory? EJIFCC. 2022 Apr:33(1):23-27 [PubMed PMID: 35645692]
Level 2 (mid-level) evidenceLock RJ. My approach to internal quality control in a clinical immunology laboratory. Journal of clinical pathology. 2006 Jul:59(7):681-4 [PubMed PMID: 16803947]
Level 2 (mid-level) evidencePetersen PH, Ricós C, Stöckl D, Libeer JC, Baadenhuijsen H, Fraser C, Thienpont L. Proposed guidelines for the internal quality control of analytical results in the medical laboratory. European journal of clinical chemistry and clinical biochemistry : journal of the Forum of European Clinical Chemistry Societies. 1996 Dec:34(12):983-99 [PubMed PMID: 8986407]
Level 2 (mid-level) evidenceAbu-Siniyeh A, Al-Shehri SS. Safety in Medical Laboratories: Perception and Practice of University Students and Laboratory Workers. Applied biosafety : journal of the American Biological Safety Association. 2021 Sep:26(Suppl 1):S34-S42. doi: 10.1089/apb.20.0050. Epub 2021 Sep 13 [PubMed PMID: 36032652]
Bayot ML, Limaiem F. Biosafety Guidelines. StatPearls. 2024 Jan:(): [PubMed PMID: 30725895]
Cornish NE, Anderson NL, Arambula DG, Arduino MJ, Bryan A, Burton NC, Chen B, Dickson BA, Giri JG, Griffith NK, Pentella MA, Salerno RM, Sandhu P, Snyder JW, Tormey CA, Wagar EA, Weirich EG, Campbell S. Clinical Laboratory Biosafety Gaps: Lessons Learned from Past Outbreaks Reveal a Path to a Safer Future. Clinical microbiology reviews. 2021 Jun 16:34(3):e0012618. doi: 10.1128/CMR.00126-18. Epub 2021 Jun 9 [PubMed PMID: 34105993]