Introduction
Alkaptonuria is a rare autosomal recessive disorder that arises from a mutation of the homogentisate 1,2-dioxygenase (HGD) gene, resulting in a deficiency of the enzyme HGD.[1] This enzyme plays a role in the phenylalanine and tyrosine degradation pathways by catabolizing the tyrosine intermediate metabolite homogentisic acid (HGA) into maleylacetoacetic acid (Figure 1).[2]
Deficiency of the HGD enzyme results in the abnormal accumulation of upstream precursors HGA, tyrosine, and phenylalanine. In particular, the buildup and deposition of HGA is the major contributor to the classic manifestations and consequences of the disease, including homogentisic aciduria, ochronosis, and ochronotic osteoarthropathy.[2]
Alkaptonuria leads to dysfunctions in various tissues throughout the body; this article will broadly cover the prominent presentations and focus on ocular manifestations of the disorder in particular. Early identification and diagnosis of the disorder are imperative for management and treatment as complications can be sight-threatening, including astigmatism, central vein occlusion, glaucoma, and blindness.[3]
The first case of alkaptonuria is believed to have presented in 1500 BC as found in the Egyptian mummy Harwa. Biopsy of the body’s hip cartilage and intervertebral discs in 1979 demonstrated extensive calcification and HGA pigmentation consistent with the process of ochronosis.[4]
Cases of the disease reported in the 16 century centered around the observation of dark urine, which continued to be the identifying feature and namesake of the “black urine disease” through the 19 century. In 1859, Dr. Carl Boedeker found that adding alkali to these patients’ urine produced the appearance of dark discoloration (later discovered to be due to HGA’s reducing power). He subsequently named the disease “alkaptonuria” in reference to “alkali” in Arabic, meaning “alkali,” “alkali” in Greek, meaning “to suck up oxygen greedily in alkali,” and “alkapton” in German, meaning “a reducing compound.”[1]
Alkaptonuria is a disease that occupies a noteworthy place in the histories of biochemistry, genetics, and medicine. In 1902, it was the first medical disorder reported to adhere to the Mendelian principles of autosomal recessive inheritance by Sir Archibald Garrod. Garrod deduced that all cases of alkaptonuria reported at the time required certain genetic contributions from both parents. In other words, the spectrum of outward manifestations could all be traced back to a specific underlying genetic trait, emphasizing the impact of science and genetics on the human constitution. At the time, many debilitating diseases were already noted to be more prevalent in consanguineous marriages, but few reports addressed consanguinity from a strictly scientific perspective. By connecting alkaptonuria to a genetic basis and referring to the similar gametes passed on by consanguineous parents, Garrod provided scientific evidence for some deleterious effects of consanguineous unions.[2]
In his 1908 Croonian Lecture to the Royal College of Physicians, Garrod also later used alkaptonuria as an example to demonstrate his concept of “the inborn error of metabolism,” now a widely used term applied to genetic disorders of innate metabolic pathways such as alkaptonuria.[5]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Biochemical experiments in the 1950s established that alkaptonuria resulted from a failure to synthesize the enzyme homogentisate 1,2-dioxygenase (called “homogentisic acid oxidase” at the time), leading to its deficiency in the tyrosine metabolism pathway.[6] Subsequent genetic mapping in the early 1990s identified the gene responsible for human alkaptonuria, temporarily called the alkaptonuria (AKU) gene, to be located on chromosome 3q.[7][8]
However, scientists were still unsure whether this AKU gene served a regulatory role in homogentisic acid oxidase enzyme synthesis or encoding role for the enzyme itself. In 1996, researchers specified the gene to be the HGD gene and demonstrated that its exons code the amino acid sequence for the enzyme itself. This study also confirmed the autosomal recessive nature of the disorder, as individuals with alkaptonuria were shown to possess two mutated HGD alleles. At the same time, heterozygous carriers with only one mutated HGD did not exhibit any signs of disease.
Although the mutated alleles necessary for the disorder are most commonly derived from the inheritance of two mutations from two heterozygous parents, homozygous or compound heterozygous mutations can also accumulate in other fashions. These alternate methods include inheritance of one mutated allele accompanied by a de novo germline mutation in the other allele, a de novo somatic mutation in a mosaic parent’s gametes, and uniparental isodisomy.[9]
Over 100 alkaptonuria mutations of the HGD allele have been identified. The most common reported mutations are missense, but other mutation types such as deletions, insertions, and nonsense have also been identified. The various mutation types result in different impacts on the molecular bonds and structure of the protein enzyme. Still, ultimately the mutations all result in a complete lack of or substantially decreased homogentisate 1,2-dioxygenase function.[8]
Epidemiology
Alkaptonuria is a rare autosomal recessive disorder with a prevalence of 1 in 100,000 to 250,000 in the general population. However, higher prevalence rates can be found in certain populations and demographics due to founder effects. In Slovakia, where prevalence reaches 1 in 19,000, most alkaptonuria pedigrees can be traced back to a specific Kysuce region in the Carpathian mountains where isolated immigrant communities settled.[10]
Similarly, populations identified in the Dominican Republic, where the prevalence of the disease is also unusually high, have been attributed to a single founder mutation C120W. This particular mutation is closely tied to the Dominican population and has only been identified outside of the country in a Dominican individual living in the United States.[11]
Consanguinity, which can be considered a version of the founder effect that is even more genetically isolated, has been observed to be associated with a higher prevalence of alkaptonuria ever since the earliest discussions of the disease. Although specific statistics of alkaptonuria prevalence in consanguineous populations have not been well studied, consanguineous mating has been expected to contribute to the presence of alkaptonuria in Arab populations.[12]
Pathophysiology
The deficient homogentisate 1,2-dioxygenase enzyme in alkaptonuria is predominantly concentrated in the liver and kidney but also occurs in the small intestine, colon, and prostate.[1][6] Loss in this enzyme’s function results in an inability of tissues to convert HGA to maleylacetoacetic acid in the tyrosine catabolism pathway, resulting in the accumulation of the intermediate metabolite HGA.[11]
Extremely high levels of plasma HGA are partially excreted into the urine. Excess HGA is then oxidized to benzoquinone acetic acid (BQA), which forms melanin-like polymers leading to the hyperpigmented appearance of dark urine.[13] As HGA accumulation progresses, the pigment deposits within connective tissues, turning tissues yellow-brown or “ochre” in a process called “ochronosis.”[2]
Ochronosis produces widely variable symptoms both in location and severity, but common consequences of polymer deposition include hyperpigmentation and progressive degeneration of the structural integrity of native tissues. This results in more stiff and brittle tissues that are not able to perform normal connective tissue functions.[14]
Within ocular structures, HGA pigment can be located phagocytosed within macrophages and fibroblasts as well as clumped in extracellular globules and granules associated with connective tissue bundles.[15] The ocular manifestations of alkaptonuria are not as well studied as the renal and joint manifestations. However, a better understanding of the ocular course of the disease is important for patient outcomes on multiple fronts. Firstly, ocular changes are often some of the earliest signs of disease, so knowledge of and ability to identify characteristic ocular findings can be very beneficial in diagnosing and managing the disease. In fact, hyperpigmentation of the sclera can present before abnormalities can even be detected on cardiac echocardiography or abdominopelvic ultrasonography.[16]
Studies reviewing mean ages of onset of ochronosis estimate that ocular signs can anticipate diagnosis of alkaptonuria by about 15 years. Furthermore, scleral hyperpigmentation can be identified with gross examination without the need for testing equipment.[3] Of note, monitoring of ocular signs is especially important in regards to patient outcomes as a progressive degeneration of ocular structures has the potential to create sight-threatening complications.
History and Physical
Due to the genetic contributions to the disease, detailed personal and family histories should be collected during the initial visit. Many presentations such as dark urine can be asymptomatic and easily missed or forgotten.
Dark urine is the earliest and only manifestation of the disorder in the pediatric population, so it continues to be one of the most heavily utilized presentations in the workup and diagnosis of the disorder.[17] Twenty-one percent of individuals with alkaptonuria can be diagnosed before one year of age, and 55% of patients receive a diagnosis due to the identification of dark urine.
Ochronosis commonly presents in the third to fifth decades of life. Presentations include scleral and ear cartilage pigmentation, tendon thickening and tears, ligament tears, muscle tears, joint and bursal effusions, kidney stones, prostate stones, aortic dilatation, cardiac valve calcification and regurgitation, and coronary artery calcification.[13]
Ochronotic osteoarthropathy occurs when ochronosis is located within the hyaline articular cartilage of joints. Osteoarthropathy is the most common symptomatic complaint and is considered the most incapacitating consequence of the disorder.[16] Forty-five percent of patients receive a diagnosis of alkaptonuria due to the presence of chronic joint pain and decreased range of motion, commonly with lumbar pain as the initial complaint. Progressive degeneration and inflammation of the joint tissues result in a narrowing of joint spaces, calcification, and eventually fusion.[13]
Symptoms involving the eyes can be very beneficial in the workup and diagnosis of the disorder. Some degree of ocular involvement is believed to be present in 67 to 70% of patients with alkaptonuria, but they often go undetected as visual acuity is usually not impacted. Yellow-brown or blue-black pigment deposition in different eye parts creates the different ocular signs of alkaptonuria (Figure 2). Although vision is typically not affected by hyperpigmentation, complications resulting from pigment deposition can be sight-threatening.[18]
The most common ocular sign is secondary to pigment deposition in the sclera, creating symmetric scleral pigmentation as seen in 83% of patients. Studies have found that pigment selectively deposits primarily in locations of the sclera with preexisting degeneration or insertion of ocular tendons.[19] The pigment then further damages the connective tissue at locations of deposition. These classic scleral pigment deposits are called “Osler’s sign,” a term also commonly used in other medical conditions such as pseudohypertension and bacterial endocarditis.[20]
Another standard (75%) ocular finding that is considered pathognomonic for the disease is the appearance of “oil-drops,” resulting from pigment deposited with a spot-like appearance in the limbus. Pigment deposition in the conjunctiva creates “vermiform” or “tube-like” conjunctival hyperpigmentation. This finding is often accompanied by increased conjunctival vessel diameter due to dilated vasculature supplying areas of pigmentation.[21] These various findings can be summarized by four main appearances of hyperpigmentation which can occur in isolation or combination: vermiform, pinguecula-like, dot-like, and laminar.
Other less common ocular signs include pigment deposition that narrows the chamber angle, such as through obstruction of trabecular meshwork or fibrosis in the periphery. This deposition location can lead to elevated intraocular pressure and glaucoma.[22] Corneoscleral pigment accumulation can also disrupt corneal curvature and has been found to cause late-onset rapidly progressive astigmatism.[23]
A few select cases have reported consequences of ocular ochronosis to include acute anterior uveitis, central vein occlusion, and macular epiretinal membrane. Still, the association of these complications to the disease has not yet been established.[24][25][26]
Gross and slit-lamp examinations can be used to visualize ochronotic hyperpigmentation in the sclera, limbus, and conjunctiva. Further testing using a tonometer, keratometer, or corneal topographer may be indicated if concerns for related complications such as glaucoma and astigmatism.
Evaluation
Clinical diagnosis of alkaptonuria can be confirmed with quantitative HGA level analyses and genetic testing. Gas or liquid chromatography-mass spectrometry can be used to detect elevated levels of HGA in urine and serum. Molecular genetic testing for HGD pathogenic variants can also be used for confirmatory diagnosis and genetic counseling for family and future progeny.
After the diagnosis is made, multi-system evaluations for occult manifestations of the disease may be beneficial. This includes radiography, electrocardiogram, echocardiogram, and renal ultrasound.[27]
Treatment / Management
Treatment of alkaptonuria centers around reducing levels of HGA or BQA by targeting various steps in the tyrosine catabolism pathway. There is currently no standard treatment protocol, so a combination of management strategies is tailored to each individual patient to achieve the best possible outcomes.
Nitisinone, or NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-cyclohexane-1,3-dione), is an oral medication that inhibits the enzyme 4-hydroxyphenylpyruvatedioxygenase. This enzyme normally converts 4-hydroxyphenylpyruvic acid to HGA. Therefore, inhibition of the enzyme via nitisinone reduces the production of HGA, alleviating symptoms of alkaptonuria.[3][28] Nitisinone has been demonstrated to reduce serum and urine HGA levels by 95% over three years, improve range of motion, and decrease reported joint pain.[29][30] A known ocular side effect of nitisinone is keratopathy secondary to elevated plasma tyrosine levels, which is managed through low-tyrosine diets.(A1)
Dietary restriction of tyrosine and phenylalanine in protein has also been utilized to decrease the amount of starting precursors for the pathway. Protein restriction has been shown to significantly lower urinary excretion of HGA, with more prominent beneficial results in patients younger than 12 years of age.[31][32] Disadvantages of this treatment modality include the difficulty of sustaining a low protein diet and the nutritional deficits that such a diet may introduce.(B3)
Ascorbic acid, or vitamin C, has been found to act as an antioxidant that prevents HGA non-enzymatic conversion into BQA, reducing hyperpigmentation damage.[33] However, further research is warranted on the effects of ascorbic acid on alkaptonuria as ascorbic acid may increase HGA production in certain populations through its action as a cofactor for the enzyme 4-hydroxyphenylpyruvatedioxygenase.[34](B3)
Gene or enzyme replacement therapy that directly replaces the missing HGD enzyme is being investigated as a potential cure for alkaptonuria. While ideal, in theory, studies focusing on this therapy have reported difficulties in its implementation. Enzyme replacement therapy requires precise tissue localization and can accumulate reactive, toxic intermediates of tyrosine metabolism in the blood and tissues.
In addition to treatment targeting biochemical pathways, supportive management that addresses the most noticeable and distressing symptoms should also be discussed. Physical and occupational therapy can help maintain muscle strength, flexibility, and range of motion, while anti-inflammatory analgesics can be used for arthralgias. In more advanced degenerative arthritis, joint arthroplasty may be indicated.[14] Ophthalmologic management includes contact lenses for astigmatism and trabeculectomy for glaucoma.[22] (B3)
For glaucoma, a trial of standard medication therapy (e.g., timolol, carbonic anhydrase inhibitor, and latanoprost topical eye drops) can be utilized. Still, it may not prove effective due to the different pathogenesis of the complication.[22] Further research would be beneficial to clarify disease and complication relationships and management.(B3)
Differential Diagnosis
Due to the rarity of the disorder and asymptomatic presentations, alkaptonuria is often mistaken for a more common disease process such as osteoarthritis. An incorrect diagnosis can lead to dangerous consequences of delayed and improper medical management of the condition.
Misdiagnosis of scleral hyperpigmentation as an ocular melanosarcoma once led to enucleation of a patient’s only remaining eye. Later pathology studies diagnosed the patient’s condition as ocular ochronosis.[35]
Joint symptoms of ochronotic osteoarthropathy can also be mistaken as other joint or hypercalcification diseases such as early-onset osteoarthritis, rheumatoid arthritis, ankylosing spondylitis, calcium pyrophosphate dihydrate crystal deposition disease, and hyperparathyroidism. However, the rapidly advanced progression of calcification, involvement of small joints, variety of calcium crystal deposits, and abnormal laboratory testing set alkaptonuria apart.[26]
Prognosis
Life expectancy in individuals with alkaptonuria is not found to be affected due to the disorder itself. Nevertheless, the consequences of the disease are progressive and can result in complications that negatively impact health and quality of life.[2] Debilitating arthritis and sight-threatening ocular complications, in particular, are most concerning.
Complications
Complications of alkaptonuria include arthritis, musculoskeletal tears, kidney stones, prostate stones, cardiac vessel stenosis, and valvular diseases.[13] Known sight-threatening ocular complications secondary to disease processes include glaucoma and astigmatism.[3] Other suggested ocular complications such as acute anterior uveitis, central vein occlusion, and macular epiretinal membrane have also been reported.[24][25][26]
Deterrence and Patient Education
Alkaptonuria is a rare metabolic disorder. Thus, patients and those caring for them should be educated on the disease process and progression to make educated decisions regarding their condition. Individuals with a positive family history can be offered further testing and family planning services. Testing includes biochemical evaluation for elevated urinary HGA levels and molecular genetic evaluation that tests for HGD mutations.
Pearls and Other Issues
- Alkaptonuria is a rare metabolic disorder that has been of interest to scientific communities since the 15th century due to its genetic, biochemical, and medical characteristics.
- Dark urine and ochronotic osteoarthropathy are the most common presentations leading to diagnosis, but unnoticed ocular signs are often present early in the disease process.
- Understanding the ocular manifestations of the disease is beneficial for early diagnosis and management of the disorder.
- A combination of treatment and supportive management is available to maximize individual results.
Enhancing Healthcare Team Outcomes
Mismanagement of alkaptonuria can result in poor patient outcomes. As alkaptonuria affects various organ systems, a multi-disciplinary approach to the workup of the patient presentation can be helpful to reach the primary diagnosis. Multiple specialties may be involved in a patient’s disease management, including medical geneticists, primary care providers, internal medicine specialists, pediatricians, orthopedic surgeons, and ophthalmologists. As is commonly seen with rare diseases, therapy options for alkaptonuria can be expensive.
Communication between the healthcare team, insurance companies, and other venues of reimbursement can help patients and families continue with adequate treatment. Family education and genetic counseling are essential for the prevention, education, and identification of alkaptonuria. Early therapeutic management of the disease is recommended and has been shown to improve the condition and slow the progression of the disease.[36] [Level 1]
Media
(Click Image to Enlarge)
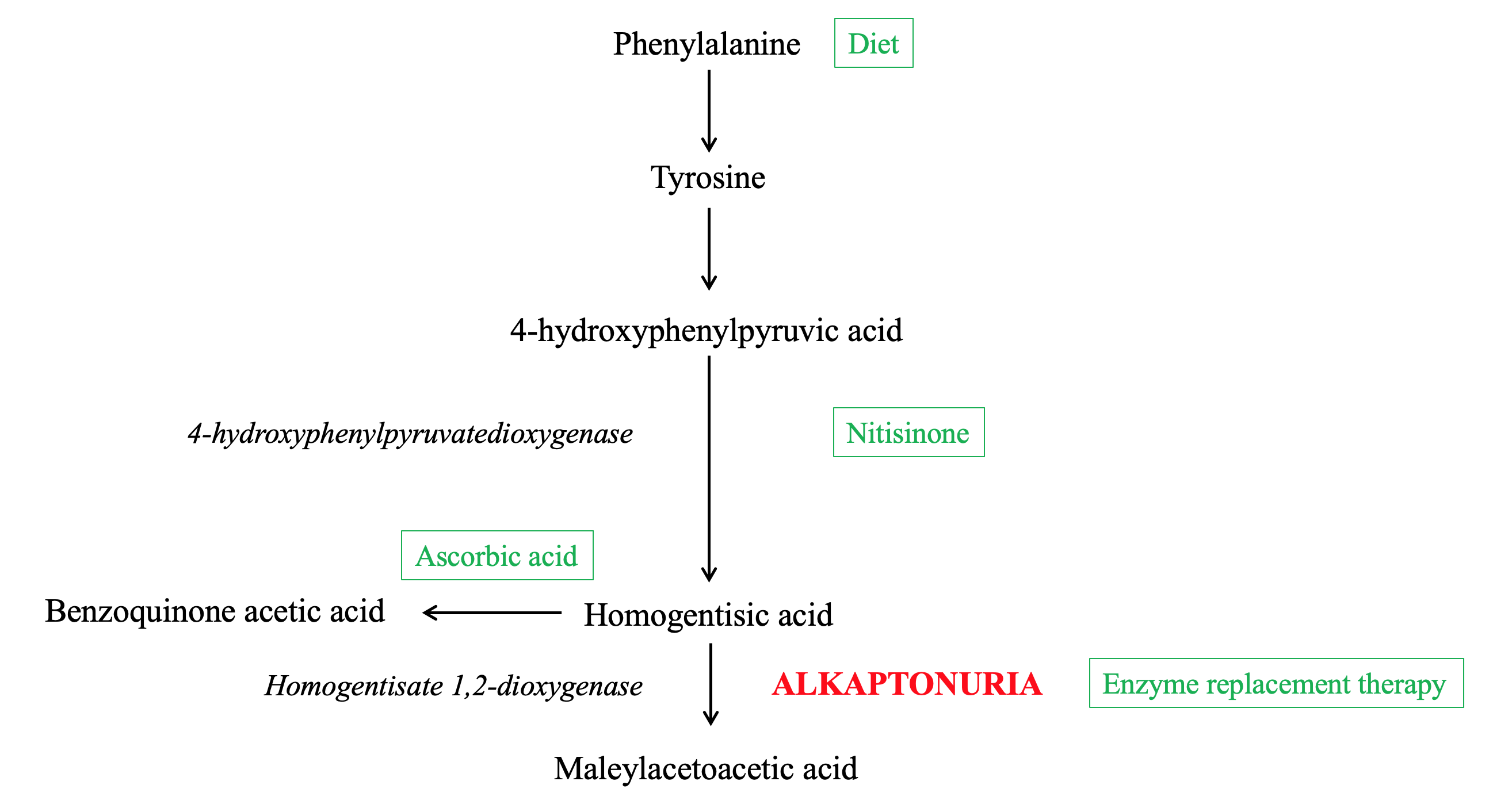
Phenylalanine catabolism pathway and steps of disease and treatment Contributed by Grace Kuang
(Click Image to Enlarge)
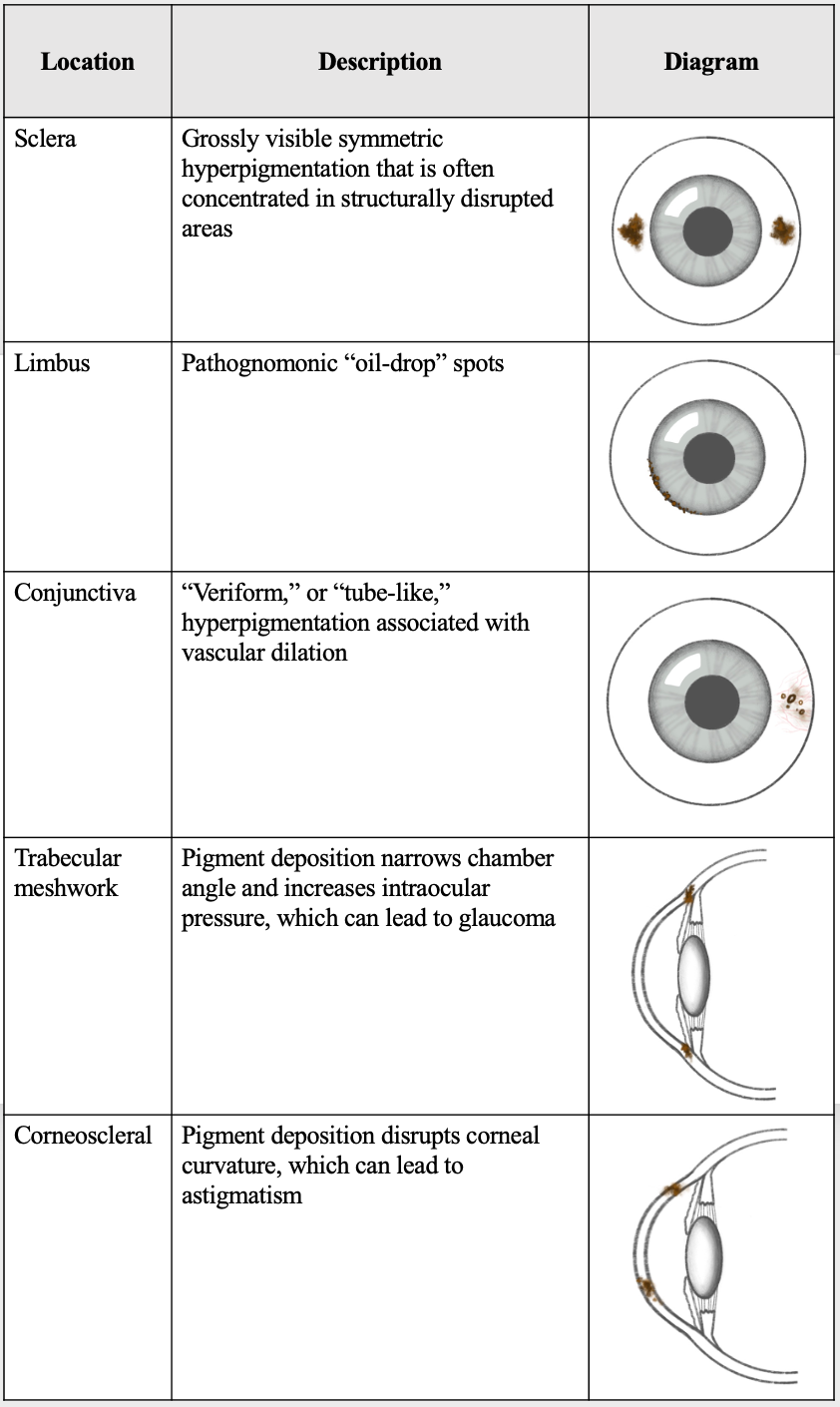
Locations of ocular ochronosis Contributed by Grace Kuang
References
Fernández-Cañón JM,Granadino B,Beltrán-Valero de Bernabé D,Renedo M,Fernández-Ruiz E,Peñalva MA,Rodríguez de Córdoba S, The molecular basis of alkaptonuria. Nature genetics. 1996 Sep; [PubMed PMID: 8782815]
Mistry JB,Bukhari M,Taylor AM, Alkaptonuria. Rare diseases (Austin, Tex.). 2013; [PubMed PMID: 25003018]
Lindner M,Bertelmann T, On the ocular findings in ochronosis: a systematic review of literature. BMC ophthalmology. 2014 Jan 30; [PubMed PMID: 24479547]
Level 1 (high-level) evidenceStenn FF,Milgram JW,Lee SL,Weigand RJ,Veis A, Biochemical identification of homogentisic acid pigment in an ochronotic egyptian mummy. Science (New York, N.Y.). 1977 Aug 5; [PubMed PMID: 327549]
Garrod AE, The incidence of alkaptonuria: a study in chemical individuality. 1902. Molecular medicine (Cambridge, Mass.). 1996 May; [PubMed PMID: 8784780]
Scriver CR, Garrod's Croonian Lectures (1908) and the charter 'Inborn Errors of Metabolism': albinism, alkaptonuria, cystinuria, and pentosuria at age 100 in 2008. Journal of inherited metabolic disease. 2008 Oct; [PubMed PMID: 18850300]
LA DU BN,ZANNONI VG,LASTER L,SEEGMILLER JE, The nature of the defect in tyrosine metabolism in alcaptonuria. The Journal of biological chemistry. 1958 Jan; [PubMed PMID: 13502394]
Janocha S,Wolz W,Srsen S,Srsnova K,Montagutelli X,Guénet JL,Grimm T,Kress W,Müller CR, The human gene for alkaptonuria (AKU) maps to chromosome 3q. Genomics. 1994 Jan 1; [PubMed PMID: 8188241]
Zatkova A, An update on molecular genetics of Alkaptonuria (AKU). Journal of inherited metabolic disease. 2011 Dec; [PubMed PMID: 21720873]
Jónsson H,Sulem P,Kehr B,Kristmundsdottir S,Zink F,Hjartarson E,Hardarson MT,Hjorleifsson KE,Eggertsson HP,Gudjonsson SA,Ward LD,Arnadottir GA,Helgason EA,Helgason H,Gylfason A,Jonasdottir A,Jonasdottir A,Rafnar T,Frigge M,Stacey SN,Th Magnusson O,Thorsteinsdottir U,Masson G,Kong A,Halldorsson BV,Helgason A,Gudbjartsson DF,Stefansson K, Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017 Sep 28; [PubMed PMID: 28959963]
Goicoechea De Jorge E,Lorda I,Gallardo ME,Pérez B,Peréz De Ferrán C,Mendoza H,Rodríguez De Córdoba S, Alkaptonuria in the Dominican Republic: identification of the founder AKU mutation and further evidence of mutation hot spots in the HGO gene. Journal of medical genetics. 2002 Jul; [PubMed PMID: 12114497]
Khalil R,Ali D,Mwafi N,Alsaraireh A,Obeidat L,Albsoul E,Al Sbou' I, Variant Analysis of Alkaptonuria Families with Significant Founder Effect in Jordan. BioMed research international. 2021; [PubMed PMID: 34235214]
Phornphutkul C,Introne WJ,Perry MB,Bernardini I,Murphey MD,Fitzpatrick DL,Anderson PD,Huizing M,Anikster Y,Gerber LH,Gahl WA, Natural history of alkaptonuria. The New England journal of medicine. 2002 Dec 26; [PubMed PMID: 12501223]
Jiang L,Cao L,Fang J,Yu X,Dai X,Miao X, Ochronotic arthritis and ochronotic Achilles tendon rupture in alkaptonuria: A 6 years follow-up case report in China. Medicine. 2019 Aug; [PubMed PMID: 31441856]
Level 3 (low-level) evidenceGaines JJ Jr, The pathology of alkaptonuric ochronosis. Human pathology. 1989 Jan; [PubMed PMID: 2643557]
Borman P,Bodur H,Ciliz D, Ochronotic arthropathy. Rheumatology international. 2002 Mar; [PubMed PMID: 11958438]
Level 3 (low-level) evidenceKisa PT,Gunduz M,Dorum S,Uzun OU,Cakar NE,Yildirim GK,Erdol S,Hismi BO,Tugsal HY,Ucar U,Gorukmez O,Gulten ZA,Kucukcongar A,Bulbul S,Sari I,Arslan N, Alkaptonuria in Turkey: Clinical and molecular characteristics of 66 patients. European journal of medical genetics. 2021 May; [PubMed PMID: 33746036]
Kampik A,Sani JN,Green WR, Ocular ochronosis. Clinicopathological, histochemical, and ultrastructural studies. Archives of ophthalmology (Chicago, Ill. : 1960). 1980 Aug; [PubMed PMID: 7417082]
Level 3 (low-level) evidenceChévez Barrios P,Font RL, Pigmented conjunctival lesions as initial manifestation of ochronosis. Archives of ophthalmology (Chicago, Ill. : 1960). 2004 Jul; [PubMed PMID: 15249376]
Level 3 (low-level) evidenceKumar A,Karthikeyan K,Vyas MT, Osler's sign revisited. Indian dermatology online journal. 2015 Jul-Aug; [PubMed PMID: 26225350]
Ben Rayana N,Chahed N,Khochtali S,Ghorbel M,Hamdi R,Rouis M,Bouajina I,Hamida FB, [Ocular ochronosis. A case report]. Journal francais d'ophtalmologie. 2008 Jun; [PubMed PMID: 18772817]
Level 3 (low-level) evidenceBacchetti S,Zeppieri M,Brusini P, A case of ocular ochronosis and chronic open-angle glaucoma: merely coincidental? Acta ophthalmologica Scandinavica. 2004 Oct; [PubMed PMID: 15453872]
Level 3 (low-level) evidenceEhongo A,Schrooyen M,Pereleux A, [Important bilateral corneal astigmatism in a case of ocular ochronosis]. Bulletin de la Societe belge d'ophtalmologie. 2005; [PubMed PMID: 15849984]
Level 3 (low-level) evidenceJohn SS,Padhan P,Mathews JV,David S, Acute anterior uveitis as the initial presentation of alkaptonuria. Journal of postgraduate medicine. 2009 Jan-Mar; [PubMed PMID: 19242077]
Level 3 (low-level) evidenceASHTON N,KIRKER JG,LAVERY FS, OCULAR FINDINGS IN A CASE OF HEREDITARY OCHRONOSIS. The British journal of ophthalmology. 1964 Aug; [PubMed PMID: 14205517]
Level 3 (low-level) evidenceDamarla N,Linga P,Goyal M,Tadisina SR,Reddy GS,Bommisetti H, Alkaptonuria: A case report. Indian journal of ophthalmology. 2017 Jun; [PubMed PMID: 28643719]
Level 3 (low-level) evidencePandey R,Kumar A,Garg R,Khanna P,Darlong V, Perioperative management of patient with alkaptonuria and associated multiple comorbidities. Journal of anaesthesiology, clinical pharmacology. 2011 Apr [PubMed PMID: 21772695]
Level 3 (low-level) evidenceAnikster Y,Nyhan WL,Gahl WA, NTBC and alkaptonuria. American journal of human genetics. 1998 Sep; [PubMed PMID: 9718357]
Level 3 (low-level) evidenceIntrone WJ,Perry MB,Troendle J,Tsilou E,Kayser MA,Suwannarat P,O'Brien KE,Bryant J,Sachdev V,Reynolds JC,Moylan E,Bernardini I,Gahl WA, A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Molecular genetics and metabolism. 2011 Aug; [PubMed PMID: 21620748]
Level 1 (high-level) evidenceSuwannarat P,O'Brien K,Perry MB,Sebring N,Bernardini I,Kaiser-Kupfer MI,Rubin BI,Tsilou E,Gerber LH,Gahl WA, Use of nitisinone in patients with alkaptonuria. Metabolism: clinical and experimental. 2005 Jun; [PubMed PMID: 15931605]
de Haas V,Carbasius Weber EC,de Klerk JB,Bakker HD,Smit GP,Huijbers WA,Duran M,Poll-The BT, The success of dietary protein restriction in alkaptonuria patients is age-dependent. Journal of inherited metabolic disease. 1998 Dec; [PubMed PMID: 9870204]
Morava E,Kosztolányi G,Engelke UF,Wevers RA, Reversal of clinical symptoms and radiographic abnormalities with protein restriction and ascorbic acid in alkaptonuria. Annals of clinical biochemistry. 2003 Jan; [PubMed PMID: 12542920]
Level 3 (low-level) evidenceKamoun P,Coude M,Forest M,Montagutelli X,Guenet JL, Ascorbic acid and alkaptonuria. European journal of pediatrics. 1992 Feb; [PubMed PMID: 1537362]
Level 3 (low-level) evidenceWolff JA,Barshop B,Nyhan WL,Leslie J,Seegmiller JE,Gruber H,Garst M,Winter S,Michals K,Matalon R, Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatric research. 1989 Aug; [PubMed PMID: 2771520]
Level 3 (low-level) evidenceSKINSNES OK, Generalized ochronosis; report of an instance in which it was misdiagnosed as melanosarcoma, with resultant enucleation of an eye. Archives of pathology. 1948 Apr; [PubMed PMID: 18891026]
Ranganath LR,Psarelli EE,Arnoux JB,Braconi D,Briggs M,Bröijersén A,Loftus N,Bygott H,Cox TF,Davison AS,Dillon JP,Fisher M,FitzGerald R,Genovese F,Glasova H,Hall AK,Hughes AT,Hughes JH,Imrich R,Jarvis JC,Khedr M,Laan D,Le Quan Sang KH,Luangrath E,Lukáčová O,Milan AM,Mistry A,Mlynáriková V,Norman BP,Olsson B,Rhodes NP,Rovenský J,Rudebeck M,Santucci A,Shweihdi E,Scott C,Sedláková J,Sireau N,Stančík R,Szamosi J,Taylor S,van Kan C,Vinjamuri S,Vrtíková E,Webb C,West E,Záňová E,Zatkova A,Gallagher JA, Efficacy and safety of once-daily nitisinone for patients with alkaptonuria (SONIA 2): an international, multicentre, open-label, randomised controlled trial. The lancet. Diabetes [PubMed PMID: 32822600]
Level 1 (high-level) evidence