Introduction
Oculo-auriculo-vertebral spectrum (OAVS) is a congenital disorder of craniofacial morphogenesis. It was first described by an ophthalmologist named Maurice Goldenhar as an association between ophthalmic, auricular, and facial features in 1952. After that, Gorlin et al. added vertebral anomalies to the list in 1963.[1] Thus, it is also called Goldenhar syndrome or facio-auriculo-vertebral syndrome, or Goldenhar-Gorlin syndrome. The term OAVS originated from 'oculoauriculovertebral dysplasia,' described by Cohen et al. in 1989.[2] They suggested that this spectrum is a phenotypic continuum with a significant overlap between malformations of structures derived from first and second branchial arches, including eyes, mouth (lips, tongue, and palate), ear, maxilla, and mandible. Internal organs like the central nervous system, heart, kidneys, and skeletal system might also be involved, due to which the term hemifacial microsomia should not be used synonymously with OAVS.[3]
Typical phenotypes include microtia, facial asymmetry, and epibulbar dermoid or lipodermoid. Tasse et al. suggested minimum diagnostic criteria to be 'either isolated microtia or preauricular tags associated with hemifacial microsomia.' [2][4]
The spectrum of OAVS can have varied clinical presentations ranging from mild or minimal facial asymmetry to severe form with marked facial defects and internal organs involvement.[2] Developmental delay along with mental retardation has also been reported in the literature.[5]
The management of patients with OAVS is challenging due to a large variety of abnormalities and differences in the severity of presentation. It requires an interprofessinoal approach and a team of specialists to plan the treatment, which should be individualized according to the presentation of each patient. This review highlights the etiology, pathogenesis, varied clinical presentations, and management options of patients with OAVS, with particular emphasis on ocular features.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiology of OAVS is multifactorial and includes both genetic and environmental factors. The presence of familial cases can explain the genetic basis of the syndrome.[3][2] Though the knowledge of the disease genetics and its pathogenesis during embryonic development has expanded, the exact causes might still be unknown.
Genetic Factors
Most commonly, OAVS is sporadic.[6][7] However, family history has been reported in 2 to 12% of cases. The phenotypic characters are similar in both familial and sporadic types of OAVS. There is a 2 to 3% risk of OAVS in the first-degree relatives of an affected individual. Various chromosomal abnormalities have been identified.[2] These include:
- The 5p15.33-pter deletion (most common type)[8][9][10]
- Deletion of 12p13.33 region involving the WNT5B gene
- Partially overlapping microduplications on 14q23.1
- Anomalies in 22q chromosome
- Anomalies in 14g32 containing GSC (GOOSECOID) gene
- Deletion of 14q31.1q31.3, a region adjacent to GSC gene
- Linkage to 15q26.2-q26.3 region
- Deletion of 12q13.33
- Partial trisomy of the 22q11 region
- Aneuploidy in chromosome X
- Translocation t(9;12) (p23;q12.2)
- Mosaicism of trisomy 7, 9 and 22
- Duplication of SIX1, SIX6, and OTX2
Environmental Factors
These include maternal or prenatal factors like diabetes mellitus during pregnancy, twin or multiple pregnancies, maternal hypothyroidism, assisted reproductive techniques, hormonal therapy, exposure to tamoxifen, celiac disease, thalidomide, embryonic blood flow disruption, vasoactive drug use, smoking, and older age group of parents.[2]
The most accepted hypothesis includes disruption of embryonic blood flow during development due to the factors mentioned above [11][12][13] Intra-natal factors include premature birth and vaginal bleeding during pregnancy.[14] Few authors have also proposed epigenetic mechanisms to explain the interaction between genetic and environmental factors.[15]
Epidemiology
OAVS has a prevalence of 1 per 3500 to 1 per 45,000 live births. Facial abnormalities are usually unilateral, leading to facial asymmetry with the right side more frequently involved. However, asymmetrical bilateral involvement (10 to 33%) can also be seen. Male infants are more commonly affected with OAVS than female infants (3 to 2). Usually, the affected children have a normal life span and normal intelligence. OAVS is most commonly sporadic, with a few familial cases (2 to 12%) reported in the literature.[4] Among familial cases, autosomal dominant as well as recessive inheritance is reported.[2] A multifactorial pattern of inheritance has been described.
Pathophysiology
It has been proposed that OAVS develops due to abnormal development of the derivatives of first and second pharyngeal arches. These pharyngeal arches made up of mesenchymal cells, develop at four weeks of gestation.[16]
Their derivatives include neural, muscular, and skeletal elements. Formation and ventrolateral migration of cranial neural crest cells are responsible for the development of craniofacial regions. Any defect in the formation, migration, proliferation, and survival of these neural crest cells is also considered an important mechanism of OAVS. Few authors have also proposed that vascular disruption leading to hematoma in the region of the ear and jaw during embryogenesis can also contribute to this spectrum.[17][18]
However, this does not explain multisystemic involvement. Another theory proposed by Lam et al. suggested that a subgroup of OAVS may be an ectodermal disorder of nondisjunction involving the otic placode leading to mesodermal disarray similar to occult spinal dysraphism (occult dysraphic syndrome, closed spinal dysraphism, or spina bifida occulta) that involves the embryonic neuraxis. OAVS may also be considered an otological disorder along with the abnormal pharyngeal arch sequence and not a true branchial arch dysplasia.[19]
Histopathology
Histopathologically, dermoid tissue consists of a stratified squamous irregularly keratinized epithelium like epidermis. The stroma is dense and contains disordered pilar structures like sebaceous glands, sweat glands, hair follicles, vascular structures, and adipose tissue.[20]
Depending upon the stromal structures, Zhong et al. have described four types of dermoid. The most common is limbal dermoid showing collagen fibers in the epidermis and dermis. Lipodermoid consists of collagen fiber tissue and adipose tissue. Complex choristoma comprises collagen fiber tissue, smooth muscle, and glandular tissue (lacrimal tissue, sweat glands, and sebaceous glands). Epibulbar osseous choristoma contains collagen fiber tissue, glandular, bone, and nervous tissue.[21]
Triolo et al. described the in vivo confocal microscopy of limbal dermoid using Heidelberg Retina Tomograph II, Cornea Module, which allows in vivo imaging of sections at different depths parallel to the scanned surface. The most superficial in vivo confocal section (18 μm depth) showed a highly regular pattern of dark gray round structures surrounded by a thin white ring, likely to be corneal epithelium. Deeper sections, at 63 μm, showed the longitudinal course of continuous, branched, tubular structures likely to be vessels.[20]
History and Physical
The phenotypic spectrum of patients with OAVS is quite heterogeneous. Multisystem involvement is quite common in OAVS. The examination findings and symptoms can be divided according to the organ system involved. The classical triad of OAVS includes mandibular hypoplasia with facial asymmetry, oculo-auricular malformations, and vertebral abnormalities.
The most common clinical features include epibulbar and lipodermoids, auricular abnormalities like microtia, hemifacial microsomia, preauricular tags, appendages and fistula, vertebral anomalies, hypoplasia of various bones like maxilla, mandible, zygomatic arch, and malar area. Mental retardation and global developmental delay have also been reported. Most common systemic malformations include ear abnormalities (100%) followed by ocular involvement (72%), vertebral (67%), central nervous system anomalies (50%), and congenital heart defects (33%).[22][23]
Central Nervous System (CNS)
Strömland et al. reported various brain abnormalities by performing neuroimaging in ten patients with OAVS. These include occipital encephalocele, absent septum pellucidum, Arnold-Chiari malformation II, cortical dysplasia, enlarged ventricles, and hypoplasia of craniofacial bones. Various functional deficits like autism spectrum disorder, developmental delay, speech abnormalities, dysphagia, hearing loss, and low vision have also been reported.
Oro-craniofacial Anomalies
These are commonly unilateral but can also have a bilateral asymmetrical presentation. The oro-facial anomalies include microsomia, macrostomia, cleft lip, cleft palate, malformation in the epiglottic fold, bifid uvula, jaw protrusion, and malocclusions. Dental abnormalities include delayed tooth development, agenesis of premolars and molars, enamel, and dentin malformations. Parents have also reported speech difficulties, feeding problems in infants, chewing and swallowing problems. Few authors have reported facial palsy, trigeminal anesthesia, and asymmetrical palatal elevation.
Auricular Abnormalities
External and middle ear abnormalities are quite common. However, the inner ear is less commonly involved. Hypoplastic external ear with preauricular tags, fistulae, low lying ear, and narrow ear canal are known. Microtia has been classified into four types by various authors depending on the severity level. Type I includes only a small external ear; type II is characterized by the moderate anomaly of the external ear such as hook-shaped, S-shaped, or question mark-shaped auricle; type III has a rudimentary external ear with no cartilage, and type IV includes anotia and absence of all external ear structures. Middle ear abnormalities include reduced cavity size and fusion between malleus and incus bones. Inner ear defect of the vestibular organ is documented. The most common type of hearing loss is conductive; however, sensorineural and combined have also been reported.
Vertebral Malformations
Spinal cord abnormalities in the cervical spine and scoliosis are reported in the literature. Renkema et al. described the different vertebral anomalies in patients with OAVS. The most commonly reported anomalies include hemivertebrae, scoliosis, and spina bifida occulta. Depending upon the location, block vertebrae, cervical ribs, hemivertebrae, and capitalization of the atlas are common in the cervical region. Hemivertebrae and block vertebrae are common in the thoracic region. Though less commonly involved, the lumbar region can present with scoliosis, hemivertebrae, and block vertebrae.[24]
Other Systemic Abnormalities
Congenital heart defects like tetralogy of Fallot, atrial and ventricular septal defects, persistent truncus arteriosus, transposition of the great vessels, aortic arch anomalies, dextrocardia, conotruncal defects, and situs inversus are common. Urogenital anomalies include renal agenesis, hydronephrosis, ectopic and fused kidneys, polycystic kidney, double and hydroureter. Gastrointestinal tract anomalies include tracheoesophageal fistula, rectal atresia, and esophageal atresia. The respiratory system can also be involved with abnormal anatomy of the larynx, pharynx, and lungs. Limb anomalies can also be seen like club foot, thumb, and radial hemimelia.
Ocular Features
The most common ocular finding is epibulbar or limbal dermoid, followed by lipodermoid and eyelid coloboma. Other findings include microphthalmia, anophthalmia, lid coloboma, ptosis, strabismus like Duane retraction syndrome, lacrimal duct stenosis. The main distinguishing feature among the ocular anomalies of Treacher Collins syndrome and OAVS is that the former has lower eyelid coloboma. In contrast, the latter has upper eyelid (junction of inner and middle one-third) coloboma.[25][26]
Rooijers et al. classified the ocular anomalies into four categories as follows[27]
- Type I: anatomical anomalies usually not impairing vision like lid coloboma, lipodermoid, orbital dystopia, lacrimal stenosis, epicanthus, and telecanthus
- Type II: anatomical anomalies likely to impair vision like epibulbar dermoid, microphthalmos, anophthalmos, exposure keratopathy, cataract, fundal coloboma, and optic nerve hypoplasia.
- Type III: motility disorders like esotropia, exotropia, Duane syndrome, abducent nerve anomaly, and ptosis.
- Type IV: refractive errors like astigmatism, anisometropia, myopia, and hyperopia.
The incidence of ocular anomalies in the literature varies from 6.7% to 100%, with severe visual impairment in 7.7 to 30%. Thus, complete ophthalmic evaluation is necessary for all patients with OAVS.[27] Various authors have also classified the common ocular features. Singh et al. classified the eyelid colobomas into small (up to 15% of eyelid length involved), medium (15 to 30%), and large (>30%), and conjunctival lipodermoid into small (up to 25% of bulbar surface involved), medium (25 to 50% of the bulbar surface), and large (>50% of the bulbar surface).
Evaluation
Early diagnosis and management are essential for preventing ocular and systemic complications. Usually, these children present with ophthalmic features like exposure keratopathy and dermoid, and the ophthalmologist should offer an interprofessional approach with complete systemic evaluation for the holistic care of the child. Early management of ear anomalies, facial deformities, and ocular anomalies also prevents social stigma in the child and leads to better mental and social development with higher self-esteem.
Complete ophthalmic evaluation is necessary at the time of presentation, including orthoptic workup. Visual acuity testing, ocular movements, strabismus evaluation, lid and adnexa examination, ocular surface, anterior and posterior segment examination, gonioscopy for angle evaluation are required. Eyelid coloboma causing exposure keratopathy is a surgical emergency and requires early correction to preserve vision.
Evaluation of Epibulbar Dermoid
Limbal dermoid is a benign congenital tumor or a choristoma which consists of normal tissue in an abnormal location, often leading to visual disturbance. They present as elevated, opaque, yellow-white mass at the limbus with hair follicles and sebaceous glands over the surface. The most common site is the inferotemporal limbus. Mann et al. initially graded the dermoids anatomically into three grades:[28]
- Grade I: superficial lesions up to 5 mm in size and localized to the limbus. These can result in oblique astigmatism and flattening of the cornea adjacent to the lesion, leading to anisometropic amblyopia.
- Grade II: larger (>5mm) lesions involving most of the cornea up to the stroma without involving Descemet’s membrane
- Grade III: large lesions involving the whole cornea and extending into the anterior chamber and iris.
Zhong et al. proposed a new clinical grading system for limbal dermoids for predicting the visual outcome after surgery.[21]
|
Parameter |
Score 0 |
Score 1 |
Score 2 |
Score 3 |
|
Area of corneal involvement |
None |
< Outer 1/4 quadrant, not involving the optical axis |
Outer 1/4 to 1/2 quadrant, not involving the optical axis |
More than 1/2 quadrant, involving the optical axis |
|
Surface shape |
None |
Slightly raised, cannot be observed when the eye is closed |
Moderately raised, can be observed when the eye is closed |
Highly raised, interferes with closing the eye |
|
Area of conjunctival involvement |
None |
<50% |
>50% |
Sclera and orbital tissue involved along with conjunctiva |
A total score of 0 to 3 classifies as grade I, 4 to 6 as grade II, and more than 6 as grade III. The authors also reported that grade I dermoids were most common, followed by grade II and III. Following surgical resection, the visual outcome was best in grade 1 dermoid with a mean logMAR (logarithm of the minimum angle of resolution) BCVA (best-corrected visual acuity) of 0.38 ± 0.05. There was a significantly worse visual outcome in grade 2 and grade 3 dermoid patients with a mean logMAR BCVA of 0.61 ± 0.09 and 0.94 ± 0.11, respectively. Zhong et al. also reported an association between clinical grading and postoperative pathology that mild pathology including dermoid and lipo-dermoid was present in large proportions of grade 1 and low clinical score dermoid whereas, complex pathology including complex choristoma and epibulbar osseous choristoma was present in high clinical score grade 2 and grade 3 dermoid patients.[21]
Ocular Investigations
Anterior segment optical coherence tomography (ASOCT) helps to look for depth of the lesion and corneal stromal involvement and planning of surgery.
Ultrasound biomicroscopy (UBM) is used to look for the depth of the lesion and the involvement of the angle of the anterior chamber.
After a complete ophthalmic evaluation, an interprofessional approach is necessary. The patient should be referred for ENT (ear nose throat), cardiology, orthopedics, pediatric assessment, anesthesia, and oro-maxillofacial examination.
Ear malformations are diagnosed and classified using CT (computed tomography) or MRI (magnetic resonance imaging) scans. Conduction and brainstem audiometry is required to assess the hearing loss. Neuropsychiatric assessment is needed to identify disturbances in intellectual functioning and the autistic spectrum. MRI and CT brain should be evaluated by a neuroradiologist to identify all CNS anomalies.
Genetic Tests and Counseling
The diagnosis of OAVS is mainly clinical, and there are no specific genetic tests for diagnosis. Moreover, a lack of clearly defined diagnostic criteria often leads to difficulty in genetic counseling. Counseling should be done for those patients who have an inherited or a de novo chromosomal abnormality. Examination of other siblings and family members should be done, and a family tree can be made to identify any inheritance pattern. Genetic counseling should be done if any other family member with similar features is identified. The risk of OAVS in a sibling in case of a proband with OAVS, with normal chromosomes and no family history, has been reported to be 2 to 3%.[2] Due to a vast phenotypic heterogenicity, it is difficult to predict the occurrence of the disease in another child.
Prenatal Diagnosis
Prenatal diagnosis is advised in families with inherited OAVS or when a previous child is born with the disease. Non-invasive investigations include fetal ultrasound to detect ear anomalies like microtia, preauricular tags, and mandibular hypoplasia in severe defects—early diagnosis of microphthalmia and orbital hypoplasia is possible with sonography even at 14 weeks of gestation. Detailed 3D scans can help to identify milder cases. Invasive diagnostics, including puncture of trophoblast or amniocentesis or chorionic villus sampling, should be advised only when genetic mutation causing OAVS is confirmed in the fetus.
Treatment / Management
The management of patients with OAVS requires an interprofessional approach starting from the first few days of life. Neonatologists and pediatricians confirm the diagnosis of congenital defects and initiate the management at the time of birth.
Following a complete evaluation, management should be tailored to the patient and focus on improving the quality of life. The management team should include a pediatrician, ophthalmologist, oral maxillofacial surgeon, audiologist, otolaryngologist, speech-language pathologist, orthodontist, plastic surgeon, radiologist, neurosurgeon, social worker, psychologist, and a geneticist. Detailed management of all systemic anomalies is outside the scope of this review.
Management of Ocular Anomalies
Upper Eyelid Coloboma
The upper eyelid coloboma is a surgical emergency and should be repaired within a few days of life to prevent exposure, corneal ulcer, perforation, corneal opacity, and amblyopia.[26] Early management includes lubricating eye drops, gels, and bandage contact lenses. Surgical management depends upon the size, location of coloboma, and general state of the child. This includes:(B3)
- Small defect (up to 25% of horizontal lid length): direct apposition
- 25 to 35% defect: Tenzel semicircular flap
- Large (>35% defect): Cutler Beard or Mustarde rotation flap, tarsomarginal grafts, and modified Hughes procedure. The risk of amblyopia due to temporary patching should be kept in mind.
Limbal Dermoid
Management of dermoid depends upon the grade (described above) and extent of the lesion as described by Mann et al. Regular follow-up is required for documenting the size of the lesion, visual acuity, ruling out amblyopia, and refraction. Medical management with spectacles is done if small lesion with mild, regular astigmatism and good vision and compliance with spectacles. Indications of surgery include poor vision or noncompliance with spectacles, enlarging lesion encroaching visual axis, amblyopia, chronic ocular surface inflammation, cosmetic reasons, anisometropia, inadequate lid closure, and all grade II and III dermoids. Surgical options include:[28]
- Grade I <50 μm thickness and <1 mm diameter: Simple excision
- Grade I <100 μm thickness and <1 mm diameter: Excision with lamellar keratectomy, amniotic membrane transplant (AMT), limbal stem cell transplant (LSCT)
- Grade II and more profound Grade I: Keratectomy, AMT, LSCT, patch graft or deep anterior lamellar keratoplasty ± AMT
- Grade III: Total anterior segment reconstruction
Surgical excision can be followed by corneal tattooing to improve the cosmetic appearance of the limbal and corneal scar. Jacob et al. described the technique of interface tattooing with SMILE donor lenticule corneal resurfacing after limbal dermoid excision. Following surgical excision, there is a risk of developing pseudopterygium over the bare area. Lang et al. described that the application of mitomycin C 0.02% for 2 minutes over the area after dermoid excision could prevent the occurrence of pseudopterygium.
Lipodermoid, more yellowish due to a deep layer of fat cells, has more extensive involvement, with the superotemporal quadrant being the commonest and often extending inferiorly. However, it rarely causes functional abnormalities like vision impairment, strabismus, or globe restriction. Thus, surgical treatment is mainly required to improve cosmesis and includes simple surgical excision and conjunctivoplasty with or without AMT.[27][26](A1)
Differential Diagnosis
Various inheritable disorders and syndromes have overlapping clinical features with OAVS.[2] However, unlike OAVS, these syndromes have distinct and recognizable phenotypic characters.
These include:
|
Syndrome |
Gene involved |
Main features |
Distinct features |
|
Treacher Collins syndrome: |
TCOF1 (5q32-q33.1) |
Zygomatic and mandible hypoplasia, external ear abnormalities, hearing impairment, preauricular hair displacement onto the cheeks, symmetrical craniofacial involvement |
Lower eyelid coloboma, absence of the lower eyelashes
|
|
Townes–Brocks syndrome |
SALL1 (16q12.1) |
Dysplastic ears (overfolded superior helices and preauricular tags), hearing impairment, thumb malformations, renal impairment |
Anal anomalies, imperforate anus |
|
CHARGE syndrome |
CHD7 (8q12.2) |
Coloboma of iris and fundus, heart malformations, choanal atresia, growth retardation, genital malformations, ear abnormalities |
The shape of the ear, semi-circular canal anomalies, choanal atresia |
|
Branchio-oto-renal spectrum disorders |
EYA1 (8q13.3), SIX5 (19q13.32), SIX1 (14q23.1) |
Malformations of the outer, middle, and inner ear, hearing impairment, branchial fistulae and cysts, renal malformations ranging from mild renal hypoplasia to bilateral renal agenesis |
branchial fistulae and cysts |
|
Phenotypic spectrum associated with mutations in EFTUD2 |
EFTUD2 (17q21.31) |
Acrofacial dysostosis, thumb anomalies, intellectual disability, zygomatic anomalies, microcephaly |
Microcephaly and esophageal atresia |
Staging
Various authors have classified OAVS in literature. Vento et al. described OMENS classification (consisting of an Orbit, Mandible, Ear, Nerve, and Soft tissue grading system). Cousley et al. and Vento et al. classified OAVS, including only the facial features, whereas Rollnick et al. did not take laterality into consideration.[29][30][31] Tasse et al. described an objective scoring system, including the laterality.[4]
|
Group |
Minimal diagnostic criteria |
Subgroups |
|
|
|
|
Unilateral |
Bilateral |
|
1 |
Microtia |
1a |
1b |
|
2 |
Preauricular tag + hemifacial microsomia |
2a |
2b |
|
3 |
Microtia/preauricular tag + hemifacial microsomia + vertebral anomalies |
3a |
3b |
Objective Scoring System
Main features/ minimal diagnostic criteria: 2 points each
Additional clinical findings: 1 point each (deafness, orofacial clefts, anomalies of the eyes, dermoid, urogenital anomalies, brain anomalies, congenital heart defects, motor and speech delay, short stature, limb anomalies, microcephaly)
Total score: 18
This scoring system can be used for an objective description of the phenotype and defines the severity of the disease, and predicts the likelihood of associated malformations in OAVS. Tasse et al. noted that the average score in bilaterally affected patients is higher than in unilateral ones. Thus, bilateral involvement is associated with more additional clinical findings. They also reported associations between specific clinical findings such as epibulbar dermoid and brain anomalies correlated with other ocular anomalies. Short stature was associated with ocular anomalies, microcephaly, limb, and vertebral anomalies. Motor developmental delay was commonly associated with speech delay, eye and brain anomalies, and short stature.[4]
Prognosis
OAVS has a varied presentation, and the prognosis depends upon the severity of the disease and timely intervention. A child can be born with life-threatening airway obstruction and other complications, making the prognosis poor. However, early intervention can prevent or manage these complications. Life expectancy is generally comparable to the normal population.[1]
Correction of all facial malformations is complex and requires multistage surgeries. Thus, it becomes difficult to predict the prognosis due to different patterns of abnormalities in different individuals. Visual prognosis is good if early correction of lid coloboma and surgical excision of dermoid is done to prevent amblyopia. Social stigma is a great concern for children's mental health due to significant facial and ear anomalies. Furthermore, school-going children facing social alienation and poor self-image can suffer from depression and feelings of isolation. Moreover, having a deformed child can affect the functioning of the whole family and might affect parents' mental health and social relations. Psychiatric consultation and counseling are essential for overcoming these problems.
The presence of a mental disability can also affect the quality of life. Thus, early management of all anatomical and functional disorders like speech abnormalities is paramount to improving the quality of life.
Complications
Ocular complications include amblyopia, corneal opacity, vision loss, exposure keratopathy, corneal ulceration, chronic ocular surface inflammation due to dermoid, and inadequate lid closure.[26]
Postoperative and Rehabilitation Care
Postoperative rehabilitation is important after surgical correction of craniofacial abnormalities and ocular anomalies. Proper refractive correction and management of amblyopia should be done after dermoid excision.
Consultations
An interprofessional approach is required for OAVS. Early consultations should be sent for timely management by the treating doctor. These include pediatrician, cardiology, oro-maxillo-facial surgeon, ophthalmology, orthodontic, otolaryngological, orthopedic, neurological, nephrology, psychiatry, and neurology. Genetic consultation is required for counseling and family planning.
Deterrence and Patient Education
Education of the family and parents is requisite at the time of diagnosis in a newborn child. Parents need to understand that it is a congenital malformation, and an interprofessional approach is required for management. Early intervention for the correction of different anomalies is important. Genetic counseling and screening of other family members are also necessary.
Enhancing Healthcare Team Outcomes
Oculo-auriculo-vertebral spectrum is a complex congenital disorder of craniofacial morphogenesis. It can affect multiple organ systems and requires interprofessional team management. Clinical presentation can vary from mild to severe form with complex phenotypic presentations. Ocular and facial anomalies should be addressed early to maximize the outcomes and reduce the social and educational impact. The treating specialist should utilize early consultation with all specialties. Speech therapy and training for the management of autistic disorders should be initiated early. Family screening and referral to a geneticist may be beneficial.
Media
(Click Image to Enlarge)
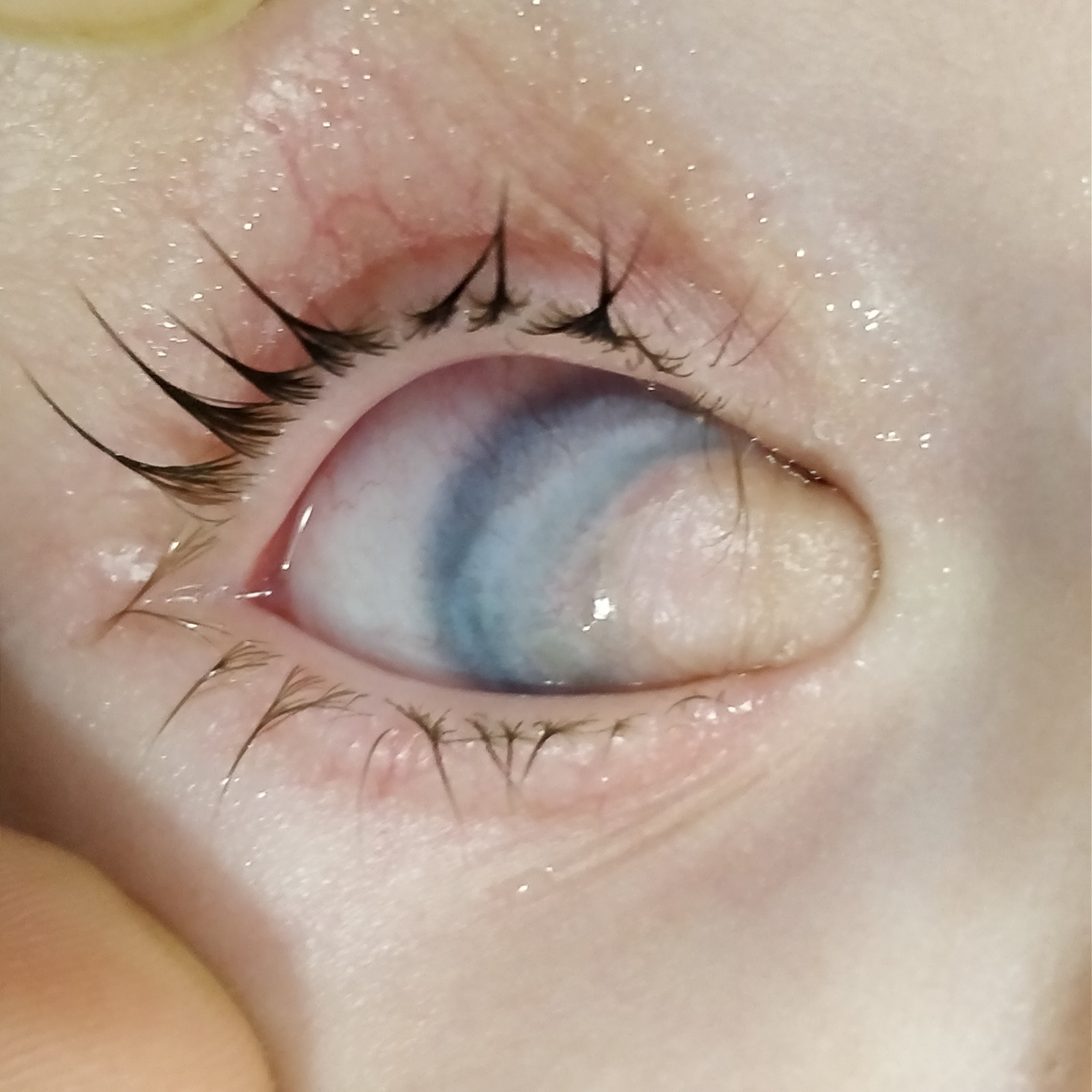
Clinical Photograph of a 18 months old patient with limbal dermoid in right eye. Contributed by Deepali Singhal, MD
References
Singh M,Kaur M,Grewal AM,Yangzes S,Yadav D,Zadeng Z,Gupta P, Ophthalmic features and management outcomes of 30 children having Goldenhar syndrome. International ophthalmology. 2020 Mar; [PubMed PMID: 31760545]
Beleza-Meireles A,Clayton-Smith J,Saraiva JM,Tassabehji M, Oculo-auriculo-vertebral spectrum: a review of the literature and genetic update. Journal of medical genetics. 2014 Oct; [PubMed PMID: 25118188]
Bogusiak K,Puch A,Arkuszewski P, Goldenhar syndrome: current perspectives. World journal of pediatrics : WJP. 2017 Oct; [PubMed PMID: 28623555]
Level 3 (low-level) evidenceTasse C,Böhringer S,Fischer S,Lüdecke HJ,Albrecht B,Horn D,Janecke A,Kling R,König R,Lorenz B,Majewski F,Maeyens E,Meinecke P,Mitulla B,Mohr C,Preischl M,Umstadt H,Kohlhase J,Gillessen-Kaesbach G,Wieczorek D, Oculo-auriculo-vertebral spectrum (OAVS): clinical evaluation and severity scoring of 53 patients and proposal for a new classification. European journal of medical genetics. 2005 Oct-Dec; [PubMed PMID: 16378924]
Strömland K,Miller M,Sjögreen L,Johansson M,Joelsson BM,Billstedt E,Gillberg C,Danielsson S,Jacobsson C,Andersson-Norinder J,Granström G, Oculo-auriculo-vertebral spectrum: associated anomalies, functional deficits and possible developmental risk factors. American journal of medical genetics. Part A. 2007 Jun 15; [PubMed PMID: 17506093]
Vendramini-Pittoli S, Kokitsu-Nakata NM. Oculoauriculovertebral spectrum: report of nine familial cases with evidence of autosomal dominant inheritance and review of the literature. Clinical dysmorphology. 2009 Apr:18(2):67-77. doi: 10.1097/MCD.0b013e328323a7dd. Epub [PubMed PMID: 19305190]
Level 3 (low-level) evidenceTug E,Atasoy HI,Koybasi Sanal S, Thrombophilia gene mutations in oculoauriculovertebral spectrum. Genetic counseling (Geneva, Switzerland). 2012 [PubMed PMID: 22611644]
Level 3 (low-level) evidenceDescartes M, Oculoauriculovertebral spectrum with 5p15.33-pter deletion. Clinical dysmorphology. 2006 Jul [PubMed PMID: 16760734]
Level 3 (low-level) evidenceJosifova DJ,Patton MA,Marks K, Oculoauriculovertebral spectrum phenotype caused by an unbalanced t(5;8)(p15.31;p23.1) rearrangement. Clinical dysmorphology. 2004 Jul; [PubMed PMID: 15194950]
Level 3 (low-level) evidenceAla-Mello S,Siggberg L,Knuutila S,von Koskull H,Taskinen M,Peippo M, Further evidence for a relationship between the 5p15 chromosome region and the oculoauriculovertebral anomaly. American journal of medical genetics. Part A. 2008 Oct 1 [PubMed PMID: 18792983]
Level 3 (low-level) evidenceRobinson LK,Hoyme HE,Edwards DK,Jones KL, Vascular pathogenesis of unilateral craniofacial defects. The Journal of pediatrics. 1987 Aug [PubMed PMID: 3612395]
Level 3 (low-level) evidenceVan Allen MI, Structural anomalies resulting from vascular disruption. Pediatric clinics of North America. 1992 Apr [PubMed PMID: 1553243]
Werler MM,Sheehan JE,Hayes C,Mitchell AA,Mulliken JB, Vasoactive exposures, vascular events, and hemifacial microsomia. Birth defects research. Part A, Clinical and molecular teratology. 2004 Jun [PubMed PMID: 15211707]
Level 2 (mid-level) evidenceRosa RF, Graziadio C, Lenhardt R, Alves RP, Paskulin GA, Zen PR. Central nervous system abnormalities in patients with oculo-auriculo-vertebral spectrum (Goldenhar syndrome). Arquivos de neuro-psiquiatria. 2010 Feb:68(1):98-102 [PubMed PMID: 20339662]
Level 2 (mid-level) evidenceFischer S,Lüdecke HJ,Wieczorek D,Böhringer S,Gillessen-Kaesbach G,Horsthemke B, Histone acetylation dependent allelic expression imbalance of BAPX1 in patients with the oculo-auriculo-vertebral spectrum. Human molecular genetics. 2006 Feb 15 [PubMed PMID: 16407370]
Szabo-Rogers HL,Smithers LE,Yakob W,Liu KJ, New directions in craniofacial morphogenesis. Developmental biology. 2010 May 1 [PubMed PMID: 19941846]
Level 3 (low-level) evidenceRichieri-Costa A,Ribeiro LA, Macrostomia, preauricular tags, and external ophthalmoplegia: a new autosomal dominant syndrome within the oculoauriculovertebral spectrum? The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial Association. 2006 Jul; [PubMed PMID: 16854200]
Level 3 (low-level) evidencePoswillo D. The pathogenesis of the first and second branchial arch syndrome. Oral surgery, oral medicine, and oral pathology. 1973 Mar:35(3):302-28 [PubMed PMID: 4631568]
Level 3 (low-level) evidenceLam CH, A theory on the embryogenesis of oculo-auriculo-vertebral (Goldenhar) syndrome. The Journal of craniofacial surgery. 2000 Nov; [PubMed PMID: 11314495]
Level 3 (low-level) evidenceTriolo G,Ferrari G,Doglioni C,Rama P, In vivo confocal microscopy in goldenhar syndrome: a case report. BMC ophthalmology. 2013 Oct 16; [PubMed PMID: 24131730]
Level 3 (low-level) evidenceZhong J,Deng Y,Zhang P,Li S,Huang H,Wang B,Zhang H,Peng L,Yang R,Xu J,Yuan J, New Grading System for Limbal Dermoid: A Retrospective Analysis of 261 Cases Over a 10-Year Period. Cornea. 2018 Jan; [PubMed PMID: 29211701]
Level 2 (mid-level) evidenceTripathy K, Sharma YR, Chawla R, Basu K, Vohra R, Venkatesh P. Triads in Ophthalmology: A Comprehensive Review. Seminars in ophthalmology. 2017:32(2):237-250. doi: 10.3109/08820538.2015.1045150. Epub 2015 Jul 6 [PubMed PMID: 26148300]
Martelli H Jr,Miranda RT,Fernandes CM,Bonan PR,Paranaíba LM,Graner E,Coletta RD, Goldenhar syndrome: clinical features with orofacial emphasis. Journal of applied oral science : revista FOB. 2010 Dec [PubMed PMID: 21308299]
Level 3 (low-level) evidenceRenkema RW,Caron CJJM,Mathijssen IMJ,Wolvius EB,Dunaway DJ,Forrest CR,Padwa BL,Koudstaal MJ, Vertebral anomalies in craniofacial microsomia: a systematic review. International journal of oral and maxillofacial surgery. 2017 Oct [PubMed PMID: 28669484]
Level 1 (high-level) evidenceRenju R, Varma BR, Kumar SJ, Kumaran P. Mandibulofacial dysostosis (Treacher Collins syndrome): A case report and review of literature. Contemporary clinical dentistry. 2014 Oct:5(4):532-4. doi: 10.4103/0976-237X.142826. Epub [PubMed PMID: 25395774]
Level 3 (low-level) evidenceSchmitzer S, Burcel M, Dăscălescu D, Popteanu IC. Goldenhar Syndrome - ophthalmologist's perspective. Romanian journal of ophthalmology. 2018 Apr-Jun:62(2):96-104 [PubMed PMID: 30206552]
Level 3 (low-level) evidenceRooijers W,Caron CJJM,Loudon SE,Padwa BL,Dunaway DJ,Forrest CR,Koudstaal MJ, Ocular and adnexal anomalies in craniofacial microsomia: a systematic review. International journal of oral and maxillofacial surgery. 2020 Sep; [PubMed PMID: 32217034]
Level 1 (high-level) evidencePirouzian A, Management of pediatric corneal limbal dermoids. Clinical ophthalmology (Auckland, N.Z.). 2013; [PubMed PMID: 23576860]
Cousley RR. A comparison of two classification systems for hemifacial microsomia. The British journal of oral & maxillofacial surgery. 1993 Apr:31(2):78-82 [PubMed PMID: 8471584]
Vento AR, LaBrie RA, Mulliken JB. The O.M.E.N.S. classification of hemifacial microsomia. The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial Association. 1991 Jan:28(1):68-76; discussion 77 [PubMed PMID: 1848447]
Level 2 (mid-level) evidenceRollnick BR, Kaye CI, Nagatoshi K, Hauck W, Martin AO. Oculoauriculovertebral dysplasia and variants: phenotypic characteristics of 294 patients. American journal of medical genetics. 1987 Feb:26(2):361-75 [PubMed PMID: 3812588]