Introduction
Glaucoma is an ocular disease characterized by ocular hypertension that leads to optic neuropathy and visual field loss.[1] Aqueous humor is secreted in the posterior chamber (PC) by the ciliary body, then moves into the anterior chamber (AC) through the pupil. The aqueous is then drained from the trabecular meshwork (TM) located at the angle between the cornea and iris. Glaucoma due to blockage of aqueous outflow from the TM by the iris or angle is defined as closed-angle glaucoma. When gonioscopy shows no evidence of angle closure, the optic neuropathy related to elevated intraocular pressure (IOP) is defined as open-angle glaucoma.
Pigment dispersion glaucoma or pigmentary glaucoma (PG) is considered secondary open-angle glaucoma. PG and pigment dispersion syndrome (PDS)[2] have similar clinical characteristics, representing a disease spectrum. PDS can convert into PG when there is the presence of elevated intraocular pressure (IOP) with visual field defects and glaucomatous optic neuropathy.[3] Friedrich E. Krukenberg described Krukenberg spindle in 1899.[4]
An interesting triad of symptoms for PG was first reported by Sugar et al. in 1949,[5] defined by corneal endothelial pigment depositions on the posterior side of the cornea, mid-peripheral radial iris transillumination defects, and heavy pigmentation in the TM.[6]
Campbell reported that pigment dispersion and accumulation in PG could be caused by pigment loss (melanin granules) from constant friction and rubbing between the posterior iris pigment epithelium and the lens zonules during physiological pupil movement,[7] which may be facilitated by the posterior bowing of the iris and reverse pupillary block that tends to be more prevalent in myopic eyes with a large iris.[8]
The pigment deposited in the AC contributes to chronic damage and death of TM cells over time, limiting the proper function and efficiency. Elevated IOP in PG results from aqueous buildup and reduction of TM aqueous humor outflow, leading to visual field loss associated with chronic optic neuropathy. The disease, which is typically bilateral, is prevalent in males with myopia and is typically diagnosed between 30 to 50 years.[9]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The principal cause of PDS and PG is the release of pigment in the anterior chamber that arises from the mechanical rubbing and friction created between the pigmented iris epithelium against the zonules and lens structures. Eyes with PDS and PG show structural findings, which tend to be more prone to irido-lenticular touch. Backward posterior bowing and reverse pupillary block have been reported as possible mechanisms for PDS and PG.[7]
Moderate myopia and deep AC tend to favor these mechanisms. It has been hypothesized that eyes with PDS may have a large iris that may bow backward and touch lens structure in the presence of a deep AC, which can normally be found in patients with myopia that have elongated eyes.[10]
In eyes at risk, physiological events like accommodation, blinking, exercise, and head movements or specific head positions can cause a reverse pupillary block, in which the irido-lenticular touch forms a flap-valve mechanism. The aqueous moves in a unidirectional manner from the PC towards the AC, but it cannot come from AC to the PC due to the valve-like mechanism. This causes high pressure within the AC, which further leads to the apposition of the iris against the anterior lens surface; this leads to elevated IOP.[9][11]
The trapped aqueous in the AC lead to posterior bowing of the peripheral iris, and the posterior pigment epithelium of the iris rubs against the zonules resulting in pigment dispersion.[9] The iris pigment epithelium may itself be more prone to shedding in PDS. Myopia may be present in 40% to 100% of the eyes with PDS. Myopia is a significant risk factor for PDS, and a higher degree of myopia may be associated with more severe PG.[12][13]
The effects of pigment showers and deposits in AC structures characterize the clinical features of PDS/PG. Krukenberg spindles are formed by pigment dispositions on the corneal endothelium. Histological studies have shown that the vertical spindle-shaped pigmented pattern on the inner side of the cornea could be due to melanin granules that undergo phagocytosis and not simply the effect of deposits on the endothelium surface.[6]
Although PDS/PG may show corneal endothelial polymegathism and pleomorphism, corneal thickness and endothelial function do not seem to be compromised in these eyes.[14] Pigment deposition can also be evident in TM when performing gonioscopy, showing homogenous and full-circumference increased pigmentation. Similar to the corneal endothelium, studies have shown that pigment is phagocytosed in TM endothelial cells.[15]
An overload of pigment in TM cells can lead to cell death and necrosis, causing collapse and alteration of trabecular beams, which leads to elevated IOP due to a decrease in the outflow.[16]
The sporadic nature of PG and low prevalence render heritability analysis difficult and limiting. Studies have reported familial aggregations in PDS/PG, showing an autosomal dominant inheritance pattern with variable or incomplete penetrance coupled with environmental factors.[17] Several genes on the long arm of chromosome 7q35-q36 were reported in 28 of 54 PDS/PG autosomal dominant patients.[18]
Recent studies based on 227 PG patients with 291 controls from Germany and the United Kingdom have shown PG may have heritable characteristics and similar genetic risks with myopia and iris pigmentation.[19] Possible genes and their chromosomal locations include LOXL1 (chromosome 15q24), MYOC (chromosome 1q25), eNOS3 (chromosome 7q35-q36), GPDS1 (chromosome 7q35-q36), and LRP1B (chromosome 2q22.1).
Pigmentary ocular hypertension (POHT) is PDS with high IOP in the absence of optic nerve head or visual field abnormality. POHT is more common in males. IOP higher than 21 mm Hg at presentation may denote a high risk of conversion to PG. Each 1 mm Hg IOP rise above 21 mm Hg may increase the chance of developing PG by 1.4 times.[20] The risk factors for conversion of PDS to PG include family history, myopia, white race, male gender, Krukenberg spindle, initial IOP of at least 21 mm Hg, and a diagnosis of PDS of more than five years.[21]
Epidemiology
Glaucoma affects over 70 million people worldwide. Primary open-angle glaucoma (POAG) is the most prevalent form of this disease, while PG accounts for about 1-1.5% in the Western world. PDS and PG primarily affect White-race individuals with myopia, showing a similar distribution between genders, yet PG shows a male predominance.[22] The male-female ratio is PG is around 3:1 (2:1 to 5:1).[23]
The low non-White prevalence could be due to the different behavior and anatomy of the iris.[24] White race iris anatomy tends to have a thinner stroma, which is more prone to posterior bowing due to less mechanical support. PG is commonly diagnosed at a relatively young age compared to POAG, with an initial diagnosis of PG in the third to fifth decade of life. PDS is an important risk factor in developing PG, with studies showing that 10% of patients with PDS develop PG after five years, which increases to 15% after 15 years.[20]
However, other studies have reported a 35 to 50% lifetime risk of developing PG in cases with PDS.[25] In the Black population, PDS is associated with hyperopia, older age, and female preponderance.[26]
Pathophysiology
Pigment showers and deposits in PDS/ PG are due to mechanical friction and rubbing between the pigmented iris epithelium and the zonules and lens structures. Eyes with PG, which tend to be bilateral, predominately male, myopic, and with a deep AC, are more prone to irido-lenticular touch, reverse pupillary block, and backward posterior bowing.[9] The dispersed pigment granules in PG can be phagocytosed in TM cells, leading to cell apoptosis, structural anatomic alterations of trabecular beams, and decreased aqueous outflow, resulting in elevated IOP and eventual chronic glaucomatous optic neuropathy.[27]
Histopathology
Corneal endothelial pigment dispositions in PDS/PG are referred to as Krukenberg spindles. It has been shown in histological studies that the vertical pigmented pattern on the cornea is made up of melanin granules phagocytosed as opposed to simple deposits on the endothelium.[6]
Pigment dispersion also reaches TM, which shows full circumference homogenous and increased pigmentation with gonioscopy. Histology studies have reported that pigment granules can also be phagocytosed by endothelial cells of the TM.[15]
The overload and burden of pigment in these cells can lead to cell necrosis, apoptosis, anatomic alterations, and poor aqueous outflow mechanisms, which can explain the increases in IOP that can lead to compression and disfunction of axons in glaucomatous optic neuropathy.[16]
History and Physical
Most forms of open-angle glaucoma are asymptomatic in the early stages. Similarly, in PG, chronic elevated IOP eventually leads to axon damage and retinal ganglion cell death with consequent glaucomatous optic neuropathy and visual field loss that affects central vision in only very late stages of the disease. Liberation of pigment in PDS/PG can be associated with IOP spikes and corneal edema, which may cause episodic symptoms of short-lasting blurred vision, eye pain, redness, photophobia, colored halos, and headaches that tend to be more frequent with extensive exercise, head movements, pupillary dilation, blinking, and prolonged reading.
Eyes with a deep AC and moderate myopia tend to favor mechanisms of pigment release due to irido-lenticular touch in PDS/PG. Posterior bowing of the iris can occur in these elongated myopic eyes at risk that tend to have a large iris and a deep AC.[10] Certain physiological events, like the ones reported above, can cause a reverse pupillary block. In these situations, aqueous is pushed from the PC to AC in a unidirectional and imbalanced fashion via a flap-valve mechanism between the iris and lens, which leads to elevated IOP.[9]
The principal signs and symptoms of PDS/ PG result from the release of pigment and pigment showers in the AC from the friction and rubbing of the posterior pigmented surface of the iris on lens structures. The clinical manifestations of PDS and PG are similar, with the latter showing a disease phase of the spectrum characterized by elevated IOP, retinal ganglion cell loss, glaucomatous optic neuropathy, and visual field defects. The classical triad of clinical signs for PG [5]evident with slit-lamp examination and gonioscopy consists of corneal endothelial pigment deposits (Krukenberg spindle), slit-like mid-peripheral radial iris transillumination defects, and homogenous heavy pigment deposition in TM.[6][28]
During a routine or emergency ophthalmic examination, patients with PDS/PG usually undergo slit-lamp examination and gonioscopy. The clinical signs related to this condition involve pigment showers that leave deposits on AC structures. Corneal epithelial edema may be noted in acute episodes of IOP. The IOP in the acute episode is usually in the twenties or thirties, but it may rarely rise to 50 mm Hg or more.
Krukenberg spindles (Figure) or pigment deposits on the corneal endothelium usually take a vertical spindle-like pattern on the central portion of the cornea. This pattern is due to the convection currents of the aqueous. The aqueous usually moves upwards near the iris and lens (posteriorly), and a downward current is seen near the corneal endothelium. Histological studies suggest melanin granules phagocytosed by endothelial cells as opposed to simple external deposits.[6]
Patients with PDS/PG can show abnormal shapes or sizes in corneal endothelium cells; however, function tends to be conserved with no signs of endotheliopathy.[14] Secondary pigment dispersion may be seen in multiple disorders, including pseudoexfoliation syndrome, after trauma, uveitis, after peripheral iridotomy, uveitis glaucoma hyphema (UGH) syndrome, and intraocular tumor, and Fuchs endothelial dystrophy. The Krukenberg spindle is not pathognomonic of PDS/PG and is not always seen in PDS. With age, the Krukenberg spindle may disappear. Factors influencing the development of the Krukenberg spindle include hormonal changes and blinking.
The anterior chamber is deep, and peripheral posterior bowing or concave appearing iris may be noted on slit-lamp examination. Floating melanin granules or pigments may be noted in the anterior chamber. These are brown-colored and smaller compared to white blood corpuscles (AC cells) which are round, white, and larger.
Iris trans-illuminating defects in a spoke-like pattern ('church window defects') in PDS/PG can easily be found by illuminating the slit-lamp light beam slightly from the temporal side through the pupil while the patient looks straight ahead. These defects are not pathognomonic of the condition but are seen in more than 85 % of cases and tend to be more detectable in light-colored eyes.[20]
The pigment loss and shower dispersion come from melanin granules that are released from the posterior iris pigment epithelium due to chronic mechanical contact and friction with the lens zonules during physiological pupil movement. Posterior bowing of the iris and reverse pupillary block can occur in myopic eyes that have deep AC with a large iris.[8][10] The transillumination defects are usually not seen in heavily pigmented irides.
The iris can show heterochromia (due to asymmetric involvement) and trough pigmentation due to the pigment deposits preferentially within the furrows on the anterior surface of the iris.[29] The melanin granules are usually seen over the inferior iris. Anisocoria can be seen in some cases due to the mechanical irritation of smooth muscles during iris contact leading to asymmetric mydriasis.[30] Histological and electron microscope studies[31] have shown cases of iris-pigmented epithelial defects, vascular hypoperfusion, and iris dilator muscle hyperplasia in PDS/PG.[32][33] Partial loss of pupillary ruff may be noted.
Dilated examination shows deposition of pigments over the zonules, on the anterior hyaloid face, and circumferentially at the equatorial region of the posterior lens capsule near the area of zonular attachment (Scheie stripe or Zentmayer line or Zentmayer ring). There may be pigments over the anterior lens surface also.
Gonioscopy can show pigment dispersion in the TM (Figure). Pigmentation tends to be full circumference and homogenous, as opposed to patchy patterns found in the TM of eyes with pseudoexfoliation glaucoma.[34] Schwalbe line can appear to be dark and more pigmented inferiorly.[35] The speckled pigmentation at or anterior to the Schwalbe line may form a Sampaolesi line. Similar to the corneal endothelium, studies have reported that the intense pigmentation in the TM of eyes with PDS/PG is due to phagocytic melanin located within TM cells.[36][37]
The pigment overload in the TM can lead to anatomic alterations, apoptosis, and cell necrosis. Structural alterations of the TM beams can lead to poorly functioning outflow mechanisms that explain the elevated IOP in PG.[38] The peripheral concavity of the iris is noted on the gonioscopy, and the angle of the anterior chamber is wide open. The TM pigmentation may decrease or disappear with age with normalization of IOP in the absence of glaucoma medications (pigment reversal sign).[39] Pigment in the inferior TM is cleared earlier than the superior angle; thus, in such cases, the superior TM appears darker than the inferior TM.
The dilated retinal examination must be done in cases with PDS/PG as these eyes may be more likely to harbor retinal lattice degeneration, and chances of rhegmatogenous retinal detachment may be more irrespective of the degree of myopia.[29] The optic nerve head should be carefully examined for changes in glaucoma. The glaucomatous damage may be asymmetric.
Evaluation
The correct diagnosis and management of patients with PDS/PG are imperative to avoid the onset and progression of functional and structural glaucomatous damage. The slit-lamp examination must include tonometry,[40] preferably with the gold standard Goldmann applanation tonometry.[41] IOP needs to be measured at baseline and every follow-up examination.[42] Patients with PDS are at risk of developing PG characterized by elevated IOP greater than 21 mmHg, in addition to visual field defects, glaucomatous optic neuropathy, and retinal nerve fiber defects. Gonioscopy is needed to confirm open angles and intense pigmentation.
Fundus examination is of utmost importance to assess optic nerve cupping and notching typically seen in glaucoma. Retinal tears and degenerations can be found in eyes with PDS/PG considering that eyes at risk tend to be myopic. Genetic testing is usually not performed for the clinical diagnosis of PDS/PG. Baseline and follow-up visual field testing are needed for the initial diagnosis of PG and the management of the disease over time. Optical coherence tomography (OCT) can provide structural information regarding peripapillary retinal nerve fiber layer (RNFL) thinning indicative of glaucomatous damage. The central corneal thickness should be performed as it affects the measurement of IOP.
Ultrasound biomicroscopy (UBM) and anterior segment OCT (AS-OCT) are used to document the concavity of the peripheral iris and the open angle of the anterior chamber. Other UBM features include posterior insertion of the iris (increased distance from scleral spur to the iris insertion); a touch of the iris with lens, zonules, and ciliary body; increased lens thickness, and increased posterior bowing of iris during accommodation.[4][43]
AS-OCT findings include increased irido-lenticular contact area, high anterior chamber volume, increased anterior chamber depth, and increased trabecular iris space area (TISA) at 500 microns and 750 microns from the scleral spur with increased angle opening distance (AOD).[4][44] Recent studies have shown that automatic refractometry could be useful for detecting and screening patients with PDS.[45]
Phenylephrine provocative test has been used to detect high risk for developing high IOP.[46] One drop of 10% phenylephrine is instilled every 5 minutes three times. Then, the pigment release in the anterior chamber is assessed.[47][48] The IOP should be checked at 1 hour and 2 hours after the installation of the drop to detect a rise in IOP and to start management of IOP if needed.
Treatment / Management
The underlying mechanical factor in PG is due to the pigment showers and deposits that get phagocytosed in TM cells leading to elevated IOP caused by reduced outflow. Chronic mechanical contact and friction between the posterior iris and lens structures lead to the release of melanin granules in PDS/PG.
Laser peripheral iridotomy (LPI) has been proposed in several studies performed in the 90s, with the aim of reducing physical contact and friction between AC structures by inducing iris flattening, reducing iris concavity, limiting posterior bowing of the iris, creating an equilibrium of the pressure in the anterior chamber and the posterior chamber; and preventing reverse pupillary block.[24][11] A study has shown that LPI may offer a protective effect against IOP rise at ten years in eyes with a high risk of pigment dispersion.[46] (A1)
The reduction of IOP may be more in patients younger than 40 years as the reverse pupillary block is aggravated by the accommodation. However, numerous studies [22] and systematic reviews have shown that LPI-treated eyes had little or no clinical differences compared to untreated eyes with PDS and that LPI cannot be considered a viable treatment in PG patients with elevated IOP.[49][50](A1)
LPI may be an option in individuals younger than 40 years, with irido-zonular contact seen on UBM and normal IOP. However, Laser treatment in PDS/PG remains debatable, thus further trials are needed to determine if different subgroups of patients with PDS/PG based on severity, AC anatomy, IOP, and other factors could benefit from LPI.
PDS patients at risk of converting to PG are advised to undergo periodic ophthalmologic examinations to monitor IOP and optic disc cupping. Similar to other forms of open-angle glaucoma, treatment is indicated for elevated IOP in the presence of functional and structural glaucomatous damage. Medical therapy, which ranges from beta-blockers, carbonic anhydrase inhibitors, prostaglandin analogs, or a combination of these, are the first-line treatment option in PG. Eye drop of pilocarpine is theoretically an ideal option for these patients considering that it reduces pupil movement, induces miosis, and limits posterior iris bowing. This therapy, however, is poorly tolerated considering the side effects including blurred vision, accommodative spasms, headaches, and retinal detachment. A randomized study found that latanoprost once daily was more effective in reducing IOP at one year and well-tolerated in patients with PG compared to timolol twice daily.[51](A1)
Eyes with PDS/PG have intense pigmentation of TM. Argon laser trabeculoplasty (ALT) has been shown to be useful in PG.[52] ALT seems to be more effective in younger patients with PG; however, effectiveness tends to diminish over time, with success rates of 45% after six years, which could be due to the high energy absorption, tendencies of overtreatment as pigmented TM absorbs more energy, and permanent tissue scarring.[53] (B2)
Older patients and patients with a long duration of PG may have increased IOP after ALT. Such eyes were not associated with more pigment dispersion to explain the IOP rise. The possible explanations include trabecular sclerosis preventing the tightening effect of ALT, and trabecular damage by ALT due to higher absorption of energy by a pigmented TM.[52]
Selective laser trabeculoplasty (SLT), which has greater absorption and effectiveness in pigmented tissues, induces less damage to TM and can be repeated over time. A study on 30 eyes undergoing 180-degree SLT revealed that the success rate at 1 year, 2 years, 3 years, and 4 years was 85%, 67%, 44%, and 14%, respectively. The average time to failure after SLT was around 27 months.[54] (B2)
A study conducted in 2005 in eyes with heavily pigmented TM reported elevated IOP after SLT; thus, caution should be used when considering this treatment option. The eyes with PG on multiple glaucoma medications, severely pigmented TM, and a previous history of ALT may be at higher risk of IOP rise after SLT.[55](B2)
Surgery can be considered in patients with failure of maximal medical therapy or laser. Trabeculectomy has been shown to be effective in patients with PG, showing comparative outcomes to POAG patients.[50] Young myopic males may be at higher risk of hypotony maculopathy and suprachoroidal hemorrhage after filtration surgery. Alternative methods like deep sclerectomy, canaloplasty,[56] and other surgical techniques with less sight-threatening operative risks can be considered for patients with uncontrolled and progressive PG.[57] Similar to other forms of open-angle glaucoma, modern surgical devices (including minimally invasive glaucoma surgery with Trabectome, iStent, Kahook dual blade) and techniques can be considered to address the decreased outflow mechanisms in PG.[58]
Differential Diagnosis
The pigment showers and fine dark deposits found in the aqueous, AC, and TM, in addition to elevated IOP, can mimic other ocular pathologies, which include:[9]
- Trauma and intraocular surgery can cause pigment deposits in the AC and TM, trans-illumination iris defects, and elevated IOP; however, history and careful slit-lamp examination can aid in the true diagnosis in these cases.
- Pseudoexfoliation syndrome (PEX) can give rise to pigment deposits in the AC and TM, elevated IOP, and trans-illumination iris defects. The typical clinical signs in PEX patients tend to differ from PDS/PG in that the deposits in the AC are white flakes as opposed to dark pigments. The pigmentation of the TM in PEX tends to be patchy and not intense and uniform. Trans-illumination iris defects in PEX are patchy, mottled, and located around the pupillary border, whereas PDS/PG eyes tend to show spoke-like mid-peripheral iris atrophy.[34]
- PG may be misdiagnosed as normal-tension glaucoma as with aging, the pigmentation reduces, and IOP may become normal.
- Intraocular tumors, like anterior uveal melanomas, can give rise to elevated IOP and pigment dispersion released by tumor cells.[59]
- Uveitis can cause a release of debris, inflammatory cells, and pigment into the AC, in addition to patchy pigmentation of the TM.[60] Trans-illumination iris defects and elevated IOP are typical in herpetic uveitis.
- Horner syndrome gives rise to anisocoria, which could be similar to eyes with PDS/PG that show asymmetric iris trans-illumination defects.[61]
- Other conditions, like rhegmatogenous retinal detachment, cataract surgery, diabetes, frequent or long-term mydriasis, Posner Schlossman syndrome, and physiological aging, can show signs of pigment dispersion in the AC and the TM, IOP spikes, and iris trans-illumination defects. A detailed history and thorough examinations are essential to provide a correct diagnosis.
Staging
Richter and colleagues suggested that there are four clinical groups of patients with PDS/PG:
- Inactive pigment dispersion and stable IOP
- Clinically visible active dispersion of pigments and stable IOP
- Active pigment dispersion, progression of glaucoma, high IOP
- Inactive pigment dispersion, progression of glaucoma, and optic nerve head damage[3]
Three clinical stages of PDS/PG have been proposed:
- Asymptomatic pigment dispersion phase that starts in early adulthood: The pigment dispersion is due to multiple precipitating factors including emotional stress, pharmacological mydriasis, and exercise. Though there is pigment release, the TM can filter these, and high IOP is usually not noted.
- The TM meshwork becomes pigmented, and high IOP is seen. The changes of glaucoma, including optic nerve head damage and visual field changes, are noted.
- The pigment clears from the TM with aging, and IOP begins to normalize. The inferior TM is less pigmented than the superior TM (pigment reversal sign).[4]
Prognosis
PDS represents the initial phase and most important risk factor (ranging from 35 to 50%) in the development of PG. The diagnosis of PG occurs more frequently in male Caucasians with myopia aged 20-50 years. Unlike most diseases, PG prognosis tends to get better with aging, in which a burn-out phase causes a reduction of pigment dispersion, clearance of pigmentation in the angle structures, increased facility of outflow, and normalization of IOP requiring less anti-glaucoma medication.[50]
The reversal of pigmentary clinical signs and normal levels of IOP in older patients can lead to the misdiagnosis of POAG or normal-tension glaucoma.[62] Patients with PG may be more likely to be steroid responders. With the increase in age, the signs of PDS may decrease due to the growth of the crystalline lens that induces a physiologic pupillary block and forward movement of the iris. The pigmentation of the TM, corneal endothelium, and over the iris may also reduce with time. The accommodation may be affected.
Complications
Eyes with PDS are at risk factor and can become PG in the presence of elevated IOP with functional and structural glaucomatous damage. These patients need to be managed similarly to other forms of open-angle glaucoma to avoid future complications and progression.
Deterrence and Patient Education
All patients should undergo routine eye examinations, which is particularly important in patients with risk factors for developing ocular hypertension and glaucoma. Visual field testing and optical coherence tomography (OCT) play an essential role in the diagnosis and management. Patients with an initial diagnosis of PDS and PG need a more stringent follow-up to monitor and control IOP. Proper education regarding therapy and adherence is essential to limit and prevent the progression of glaucomatous damage. Treatment with laser and surgery can be considered in select patients that show uncontrolled IOP with maximum local therapy and glaucomatous progression.
Enhancing Healthcare Team Outcomes
Undiagnosed PG can lead to irreversible functional visual field loss, glaucomatous optic neuropathy, and progressive retinal nerve layer thinning, with possible central visual loss arising in the late stages of the disease. Early diagnosis in patients with PDS can help identify patients at risk of developing elevated IOP that may require more stringent management. The ophthalmologist needs to collaborate with other healthcare professionals within the hospital and community setting.
Routine eye examinations that include tonometry, slit-lamp examination, and fundus examination, coupled with visual field testing and optical coherence tomography (OCT), can provide the proper tools to manage patients with PDS at risk of developing elevated IOP. In patients on therapy for PG, primary and secondary care is necessary to ensure periodic assessments to control and limit glaucoma progression.[Level 5]
Media
(Click Image to Enlarge)
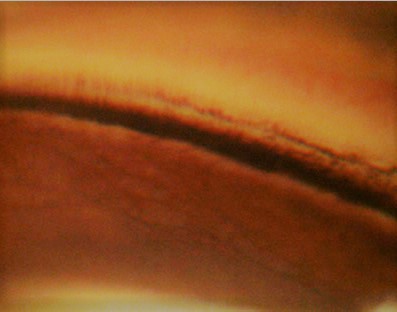
In Pigment Dispersion Syndrome (PDS) and/or Pigmentary Glaucoma (PG), pigment deposition can also be evident in TM when preforming gonioscopy, showing homogenous and full circumference increased pigmentation. Contributed by Marco Zeppieri, MD, PhD. Image courtesy of Paolo Brusini, MD.
(Click Image to Enlarge)
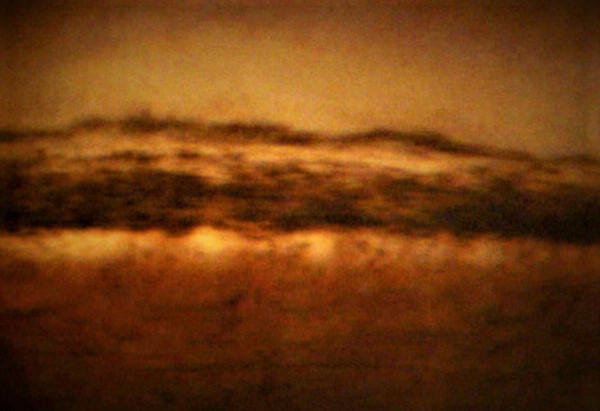
Increased pigmentation in the trabecular meshwork when preforming gonioscopy in Pigment Dispersion Syndrome (PDS) and/or Pigmentary Glaucoma (PG) . Contributed by Marco Zeppieri, MD, PhD. Image courtesy of Paolo Brusini, MD.
(Click Image to Enlarge)
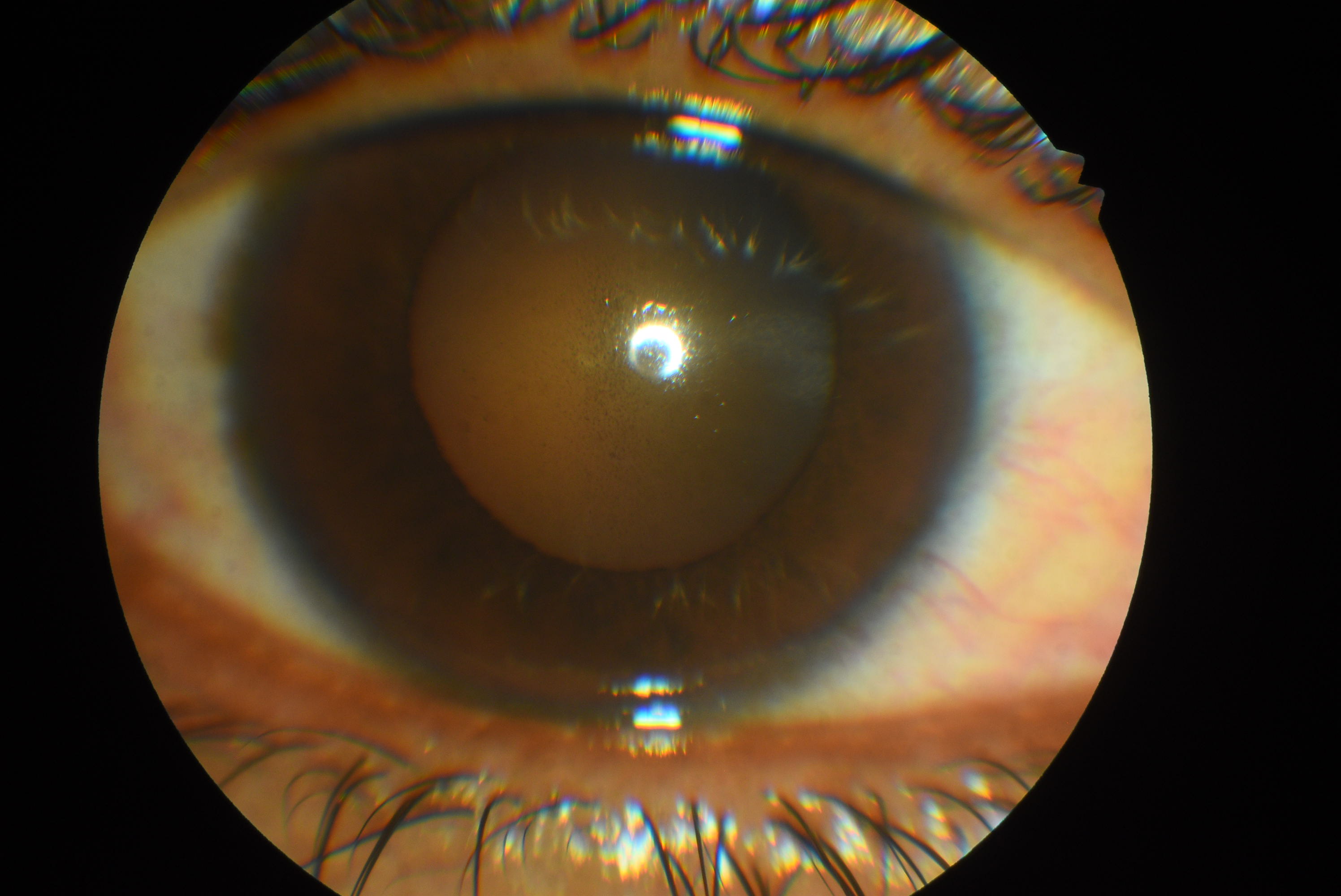
Krukenberg spindle Contributed by Dr. Koushik Tripathy
(Click Image to Enlarge)
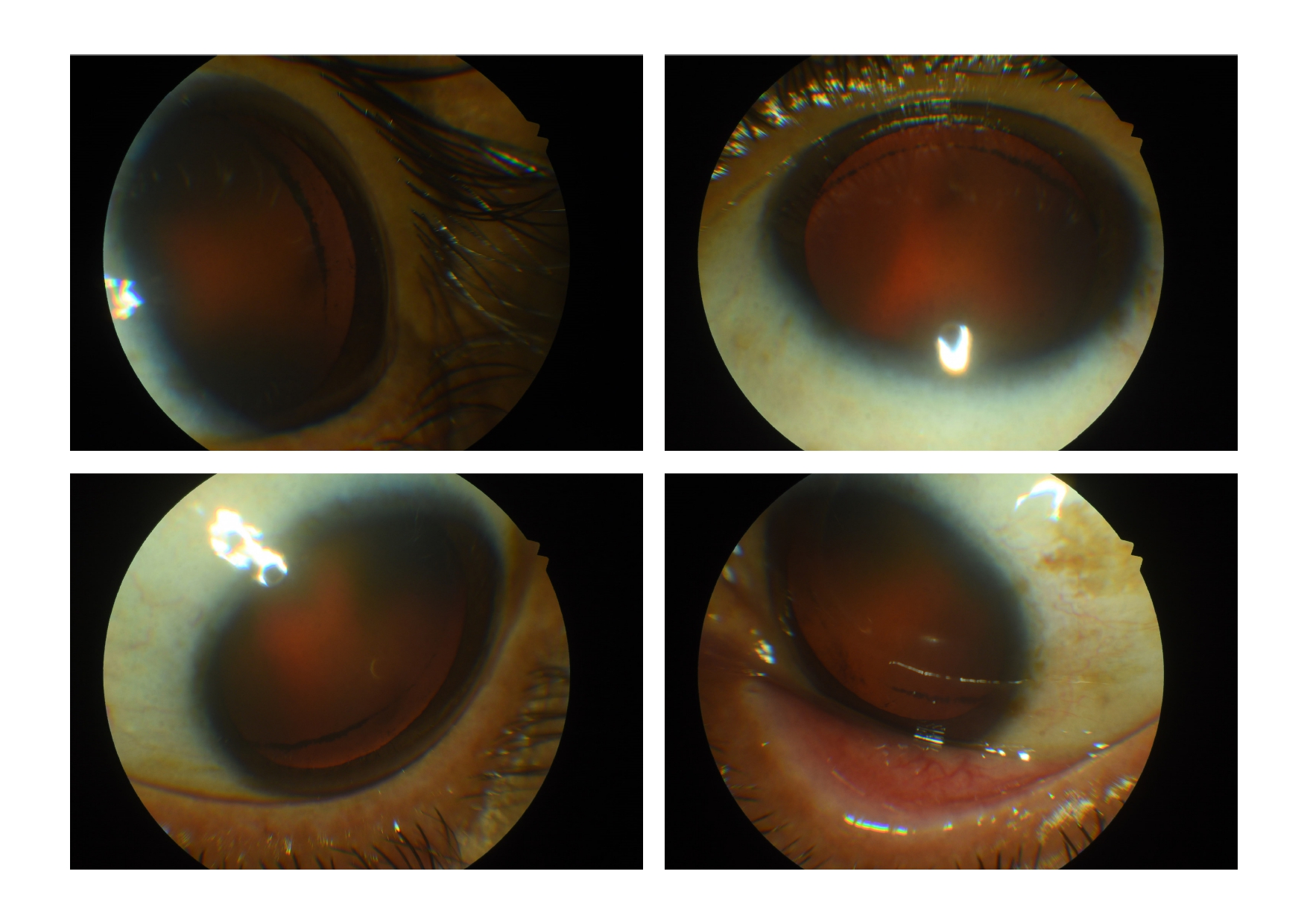
Zentmayer line (Schie line) in an eye with pigment dispersion syndrome Contributed by Koushik Tripathy, MD
References
Mahabadi N, Foris LA, Tripathy K. Open Angle Glaucoma. StatPearls. 2024 Jan:(): [PubMed PMID: 28722917]
Gurnani B, Kaur K. Pigment Dispersion Syndrome. StatPearls. 2024 Jan:(): [PubMed PMID: 35593834]
Okafor K, Vinod K, Gedde SJ. Update on pigment dispersion syndrome and pigmentary glaucoma. Current opinion in ophthalmology. 2017 Mar:28(2):154-160. doi: 10.1097/ICU.0000000000000352. Epub [PubMed PMID: 27898469]
Level 3 (low-level) evidenceBustamante-Arias A, Ruiz-Lozano RE, Carlos Alvarez-Guzman J, Gonzalez-Godinez S, Rodriguez-Garcia A. Pigment dispersion syndrome and its implications for glaucoma. Survey of ophthalmology. 2021 Sep-Oct:66(5):743-760. doi: 10.1016/j.survophthal.2021.01.002. Epub 2021 Jan 12 [PubMed PMID: 33444629]
Level 3 (low-level) evidenceSUGAR HS, BARBOUR FA. Pigmentary glaucoma; a rare clinical entity. American journal of ophthalmology. 1949 Jan:32(1):90-2 [PubMed PMID: 18122917]
Sugar HS. Pigmentary glaucoma. A 25-year review. American journal of ophthalmology. 1966 Sep:62(3):499-507 [PubMed PMID: 5920432]
Campbell DG. Pigmentary dispersion and glaucoma. A new theory. Archives of ophthalmology (Chicago, Ill. : 1960). 1979 Sep:97(9):1667-72 [PubMed PMID: 475638]
Level 3 (low-level) evidencePotash SD, Tello C, Liebmann J, Ritch R. Ultrasound biomicroscopy in pigment dispersion syndrome. Ophthalmology. 1994 Feb:101(2):332-9 [PubMed PMID: 8115154]
Niyadurupola N, Broadway DC. Pigment dispersion syndrome and pigmentary glaucoma--a major review. Clinical & experimental ophthalmology. 2008 Dec:36(9):868-82. doi: 10.1111/j.1442-9071.2009.01920.x. Epub [PubMed PMID: 19278484]
Ritch R. A unification hypothesis of pigment dispersion syndrome. Transactions of the American Ophthalmological Society. 1996:94():381-405; discussion 405-9 [PubMed PMID: 8981706]
Level 3 (low-level) evidenceKarickhoff JR. Pigmentary dispersion syndrome and pigmentary glaucoma: a new mechanism concept, a new treatment, and a new technique. Ophthalmic surgery. 1992 Apr:23(4):269-77 [PubMed PMID: 1589198]
Level 3 (low-level) evidenceFarrar SM, Shields MB, Miller KN, Stoup CM. Risk factors for the development and severity of glaucoma in the pigment dispersion syndrome. American journal of ophthalmology. 1989 Sep 15:108(3):223-9 [PubMed PMID: 2774030]
Level 2 (mid-level) evidenceGramer E, Thiele H, Ritch R. [Family history of glaucoma and risk factors in pigmentary glaucoma. A new clinical study]. Klinische Monatsblatter fur Augenheilkunde. 1998 Jun:212(6):454-64 [PubMed PMID: 9715466]
Lehto I, Ruusuvaara P, Setälä K. Corneal endothelium in pigmentary glaucoma and pigment dispersion syndrome. Acta ophthalmologica. 1990 Dec:68(6):703-9 [PubMed PMID: 2080703]
Shimizu T,Hara K,Futa R, Fine structure of trabecular meshwork and iris in pigmentary glaucoma. Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. Albrecht von Graefe's archive for clinical and experimental ophthalmology. 1981; [PubMed PMID: 6908466]
Level 3 (low-level) evidenceAlvarado JA, Murphy CG. Outflow obstruction in pigmentary and primary open angle glaucoma. Archives of ophthalmology (Chicago, Ill. : 1960). 1992 Dec:110(12):1769-78 [PubMed PMID: 1463421]
Lascaratos G, Shah A, Garway-Heath DF. The genetics of pigment dispersion syndrome and pigmentary glaucoma. Survey of ophthalmology. 2013 Mar-Apr:58(2):164-75. doi: 10.1016/j.survophthal.2012.08.002. Epub 2012 Dec 6 [PubMed PMID: 23218808]
Level 3 (low-level) evidenceAndersen JS, Pralea AM, DelBono EA, Haines JL, Gorin MB, Schuman JS, Mattox CG, Wiggs JL. A gene responsible for the pigment dispersion syndrome maps to chromosome 7q35-q36. Archives of ophthalmology (Chicago, Ill. : 1960). 1997 Mar:115(3):384-8 [PubMed PMID: 9076212]
Simcoe MJ, Weisschuh N, Wissinger B, Hysi PG, Hammond CJ. Genetic Heritability of Pigmentary Glaucoma and Associations With Other Eye Phenotypes. JAMA ophthalmology. 2020 Mar 1:138(3):294-299. doi: 10.1001/jamaophthalmol.2019.5961. Epub [PubMed PMID: 31999318]
Siddiqui Y, Ten Hulzen RD, Cameron JD, Hodge DO, Johnson DH. What is the risk of developing pigmentary glaucoma from pigment dispersion syndrome? American journal of ophthalmology. 2003 Jun:135(6):794-9 [PubMed PMID: 12788118]
Level 2 (mid-level) evidenceZeppieri M. Pigment dispersion syndrome: A brief overview. Journal of clinical and translational research. 2022 Oct 31:8(5):344-350 [PubMed PMID: 36518550]
Level 3 (low-level) evidenceMichelessi M, Lindsley K. Peripheral iridotomy for pigmentary glaucoma. The Cochrane database of systematic reviews. 2016 Feb 12:2(2):CD005655. doi: 10.1002/14651858.CD005655.pub2. Epub 2016 Feb 12 [PubMed PMID: 26871761]
Level 1 (high-level) evidenceDi Pippo M, Ciancimino C, Scuderi L, Perdicchi A. An Iconic Case of Pigmentary Glaucoma: Brief Review of the Literature. Case reports in ophthalmology. 2020 May-Aug:11(2):377-384. doi: 10.1159/000508605. Epub 2020 Jul 20 [PubMed PMID: 32884553]
Level 3 (low-level) evidenceYang JW, Sakiyalak D, Krupin T. Pigmentary glaucoma. Journal of glaucoma. 2001 Oct:10(5 Suppl 1):S30-2 [PubMed PMID: 11890269]
Ritch R, Mudumbai R, Liebmann JM. Combined exfoliation and pigment dispersion: paradigm of an overlap syndrome. Ophthalmology. 2000 May:107(5):1004-8 [PubMed PMID: 10811097]
Level 2 (mid-level) evidenceSemple HC, Ball SF. Pigmentary glaucoma in the black population. American journal of ophthalmology. 1990 May 15:109(5):518-22 [PubMed PMID: 2333915]
Wang C, Dang Y, Loewen RT, Waxman S, Shah P, Xia X, Loewen NA. Impact of pigment dispersion on trabecular meshwork cells. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2019 Jun:257(6):1217-1230. doi: 10.1007/s00417-019-04300-7. Epub 2019 Mar 28 [PubMed PMID: 30919079]
Qing G, Wang N, Tang X, Zhang S, Chen H. Clinical characteristics of pigment dispersion syndrome in Chinese patients. Eye (London, England). 2009 Aug:23(8):1641-6. doi: 10.1038/eye.2008.328. Epub 2008 Nov 7 [PubMed PMID: 18989347]
Level 2 (mid-level) evidenceScheie HG, Cameron JD. Pigment dispersion syndrome: a clinical study. The British journal of ophthalmology. 1981 Apr:65(4):264-9 [PubMed PMID: 7236571]
Feibel RM, Perlmutter JC. Anisocoria in the pigmentary dispersion syndrome. American journal of ophthalmology. 1990 Dec 15:110(6):657-60 [PubMed PMID: 2248330]
Level 3 (low-level) evidenceHaynes WL, Alward WL, Thompson HS. Distortion of the pupil in patients with the pigment dispersion syndrome. Journal of glaucoma. 1994 Winter:3(4):329-32 [PubMed PMID: 19920618]
Fine BS, Yanoff M, Scheie HG. Pigmentary "glaucoma". A histologic study. Transactions - American Academy of Ophthalmology and Otolaryngology. American Academy of Ophthalmology and Otolaryngology. 1974 Mar-Apr:78(2):OP314-25 [PubMed PMID: 4825061]
Rodrigues MM, Spaeth GL, Weinreb S, Sivalingam E. Spectrum of trabecular pigmentation in open-angle glaucoma: a clinicopathologic study. Transactions. Section on Ophthalmology. American Academy of Ophthalmology and Otolaryngology. 1976 Mar-Apr:81(2):258-76 [PubMed PMID: 936399]
Level 3 (low-level) evidencePrince AM, Ritch R. Clinical signs of the pseudoexfoliation syndrome. Ophthalmology. 1986 Jun:93(6):803-7 [PubMed PMID: 3737125]
Petersen HP. Can pigmentary deposits on the trabecular meshwork increase the resistance of the aqueous outflow? Acta ophthalmologica. 1969:47(3):743-9 [PubMed PMID: 5394638]
Richardson TM, Hutchinson BT, Grant WM. The outflow tract in pigmentary glaucoma: a light and electron microscopic study. Archives of ophthalmology (Chicago, Ill. : 1960). 1977 Jun:95(6):1015-25 [PubMed PMID: 869744]
Level 3 (low-level) evidenceSokol J, Stegman Z, Liebmann JM, Ritch R. Location of the iris insertion in pigment dispersion syndrome. Ophthalmology. 1996 Feb:103(2):289-93 [PubMed PMID: 8594516]
Gottanka J, Johnson DH, Grehn F, Lütjen-Drecoll E. Histologic findings in pigment dispersion syndrome and pigmentary glaucoma. Journal of glaucoma. 2006 Apr:15(2):142-51 [PubMed PMID: 16633228]
Level 3 (low-level) evidenceSpeakman JS. Pigmentary dispersion. The British journal of ophthalmology. 1981 Apr:65(4):249-51 [PubMed PMID: 7236568]
Level 3 (low-level) evidenceBrusini P, Salvetat ML, Zeppieri M. It Is All about Pressure. Journal of clinical medicine. 2022 Jun 23:11(13):. doi: 10.3390/jcm11133640. Epub 2022 Jun 23 [PubMed PMID: 35806926]
Brusini P, Salvetat ML, Zeppieri M. How to Measure Intraocular Pressure: An Updated Review of Various Tonometers. Journal of clinical medicine. 2021 Aug 27:10(17):. doi: 10.3390/jcm10173860. Epub 2021 Aug 27 [PubMed PMID: 34501306]
Salvetat ML, Zeppieri M, Tosoni C, Brusini P, Medscape. Baseline factors predicting the risk of conversion from ocular hypertension to primary open-angle glaucoma during a 10-year follow-up. Eye (London, England). 2016 Jun:30(6):784-95. doi: 10.1038/eye.2016.86. Epub 2016 May 13 [PubMed PMID: 27174381]
Adam RS, Pavlin CJ, Ulanski LJ. Ultrasound biomicroscopic analysis of iris profile changes with accommodation in pigmentary glaucoma and relationship to age. American journal of ophthalmology. 2004 Oct:138(4):652-4 [PubMed PMID: 15488798]
Birner B, Tourtas T, Wessel JM, Jünemann AG, Mardin CY, Kruse FE, Laemmer R. [Pigment dispersion syndrome and pigmentary glaucoma. Morphometric analysis of the anterior chamber segment with SL-OCT]. Der Ophthalmologe : Zeitschrift der Deutschen Ophthalmologischen Gesellschaft. 2014:111(7):638-43. doi: 10.1007/s00347-013-2943-6. Epub [PubMed PMID: 24062148]
Level 1 (high-level) evidenceBrusini P, Papa V. Use of an automatic refractometer as a screening tool for pigment dispersion syndrome detection. Survey of ophthalmology. 2022 Nov-Dec:67(6):1723. doi: 10.1016/j.survophthal.2022.07.003. Epub 2022 Jul 17 [PubMed PMID: 35858664]
Level 3 (low-level) evidenceGandolfi SA, Ungaro N, Tardini MG, Ghirardini S, Carta A, Mora P. A 10-year follow-up to determine the effect of YAG laser iridotomy on the natural history of pigment dispersion syndrome: a randomized clinical trial. JAMA ophthalmology. 2014 Dec:132(12):1433-8. doi: 10.1001/jamaophthalmol.2014.3291. Epub [PubMed PMID: 25188224]
Level 1 (high-level) evidenceMITSUI Y, TAKAGI Y. Nature of aqueous floaters due to sympathomimetic mydriatics. Archives of ophthalmology (Chicago, Ill. : 1960). 1961 May:65():626-31 [PubMed PMID: 13771427]
Epstein DL, Boger WP 3rd, Grant WM. Phenylephrine provocative testing in the pigmentary dispersion syndrome. American journal of ophthalmology. 1978 Jan:85(1):43-50 [PubMed PMID: 619685]
Level 3 (low-level) evidenceJampel HD. Lack of effect of peripheral laser iridotomy in pigment dispersion syndrome. Archives of ophthalmology (Chicago, Ill. : 1960). 1993 Dec:111(12):1606 [PubMed PMID: 8155025]
Level 3 (low-level) evidenceMigliazzo CV, Shaffer RN, Nykin R, Magee S. Long-term analysis of pigmentary dispersion syndrome and pigmentary glaucoma. Ophthalmology. 1986 Dec:93(12):1528-36 [PubMed PMID: 3808615]
Mastropasqua L,Carpineto P,Ciancaglini M,Gallenga PE, A 12-month, randomized, double-masked study comparing latanoprost with timolol in pigmentary glaucoma. Ophthalmology. 1999 Mar [PubMed PMID: 10080213]
Level 1 (high-level) evidenceLunde MW. Argon laser trabeculoplasty in pigmentary dispersion syndrome with glaucoma. American journal of ophthalmology. 1983 Dec:96(6):721-5 [PubMed PMID: 6660259]
Ritch R, Liebmann J, Robin A, Pollack IP, Harrison R, Levene RZ, Hagadus J. Argon laser trabeculoplasty in pigmentary glaucoma. Ophthalmology. 1993 Jun:100(6):909-13 [PubMed PMID: 8510905]
Level 2 (mid-level) evidenceAyala M. Long-term outcomes of selective laser trabeculoplasty (SLT) treatment in pigmentary glaucoma patients. Journal of glaucoma. 2014 Dec:23(9):616-9. doi: 10.1097/IJG.0b013e318287abb7. Epub [PubMed PMID: 23429632]
Level 2 (mid-level) evidenceHarasymowycz PJ, Papamatheakis DG, Latina M, De Leon M, Lesk MR, Damji KF. Selective laser trabeculoplasty (SLT) complicated by intraocular pressure elevation in eyes with heavily pigmented trabecular meshworks. American journal of ophthalmology. 2005 Jun:139(6):1110-3 [PubMed PMID: 15953448]
Level 2 (mid-level) evidenceBrusini P, Papa V, Zeppieri M. Canaloplasty in Pseudoexfoliation Glaucoma. Can It Still Be Considered a Good Choice? Journal of clinical medicine. 2022 Apr 30:11(9):. doi: 10.3390/jcm11092532. Epub 2022 Apr 30 [PubMed PMID: 35566656]
Brusini P, Papa V. Canaloplasty in Pigmentary Glaucoma: Long-Term Outcomes and Proposal of a New Hypothesis on Its Intraocular Pressure Lowering Mechanism. Journal of clinical medicine. 2020 Dec 12:9(12):. doi: 10.3390/jcm9124024. Epub 2020 Dec 12 [PubMed PMID: 33322842]
Klamann MK, Gonnermann J, Pahlitzsch M, Maier AK, Joussen AM, Torun N, Bertelmann E. iStent inject in phakic open angle glaucoma. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2015 Jun:253(6):941-7. doi: 10.1007/s00417-015-3014-2. Epub 2015 Apr 26 [PubMed PMID: 25912085]
Chaudhry IM, Moster MR, Augsburger JJ. Iris ring melanoma masquerading as pigmentary glaucoma. Archives of ophthalmology (Chicago, Ill. : 1960). 1997 Nov:115(11):1480-1 [PubMed PMID: 9366690]
Level 3 (low-level) evidenceDua HS, Dick AD, Watson NJ, Forrester JV. A spectrum of clinical signs in anterior uveitis. Eye (London, England). 1993:7 ( Pt 1)():68-73 [PubMed PMID: 8325427]
Level 3 (low-level) evidenceHaynes WL, Thompson HS, Kardon RH, Alward WL. Asymmetric pigmentary dispersion syndrome mimicking Horner's syndrome. American journal of ophthalmology. 1991 Oct 15:112(4):463-4 [PubMed PMID: 1928256]
Level 3 (low-level) evidenceScuderi G, Contestabile MT, Scuderi L, Librando A, Fenicia V, Rahimi S. Pigment dispersion syndrome and pigmentary glaucoma: a review and update. International ophthalmology. 2019 Jul:39(7):1651-1662. doi: 10.1007/s10792-018-0938-7. Epub 2018 May 2 [PubMed PMID: 29721842]