 Neuromyelitis Optica Spectrum Disorder (NMOSD)
Neuromyelitis Optica Spectrum Disorder (NMOSD)
Introduction
Neuromyelitis optica spectrum disorder (NMOSD), previously called neuromyelitis optica or Devic disease, was first described by Dr. Eugene Devic in 1894 while evaluating a patient with optic neuritis with accompanying neuromuscular manifestations (see Image 1. Neuromyelitis Optica). That same year, Fernand Gault, Dr. Devic’s student, published his doctoral thesis presenting a literature review of previous medical cases, including the clinicopathological findings of Dr. Devic’s case.[1] Multiple sclerosis (MS) was the most prevalent identifiable autoimmune disease causing optic neuritis for many years. Recent discoveries have highlighted rarer, more sinister autoimmune diseases manifesting with optic neuritis, such as neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein antibody disease (MOGAD).[2]
In recent years, the definition of neuromyelitis optica has been expanded as a specific antibody was discovered in the serum of affected patients, and various manifestations have been recognized in a spectrum of diseases. Because of this, the term neuromyelitis optica spectrum disorder is now used to include optic neuritis with spinal cord manifestations and other neurologic disorders associated with the serum aquaporin-4 immunoglobulin G antibodies (AQP4-IgG).[3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiology of NMOSD is still unclear. In the past, NMOSD was considered a variant of MS, but recent studies have elucidated the differences in the disease process, manifestations, and management. Whole-genome sequencing has identified some at-risk genotypes for NMOSD, and major histocompatibility complexes (including HLA-DRB1*03:01) have been identified.[4] The genetic variant of major histocompatibility complexes of NMOSD is more similar to systemic lupus erythematosus (SLE) than MS.[4]
Epidemiology
The prevalence of NMOSD is approximately 0.3 to 4.4 per 100,000 people.[5] NMOSD characteristically occurs in females (80%) and younger patients between 30 and 40 years of age.[6] Pediatric cases of NMOSD are rare but have been reported occasionally, comprising less than 5% of cases.[7] NMOSD occurs worldwide, and most information on the disorder is limited to economically advantaged countries, likely due to the relative ease of access to magnetic resonance imaging (MRI) and targeted antibody testing compared to emerging countries.[8] Worldwide, NMOSD has a variable prevalence among demyelinating diseases, comprising only 1% to 2% in the United States and Italy but 13.7% in India and over 30% in Thailand.[8]
Ethnicity appears to play a role in the phenotype of the disease. Findings from a study in Cuba showed that African-heritage patients with NMOSD were older, had more lesions on MRI, and had more relapses than comparable patients of other ethnicities.[9] The variations in the epidemiology of this disease should give the clinician additional information to consider the diagnosis of NMOSD, especially if a patient presents with a suspected demyelinating disease and is of Asian or Indian heritage or is older and of African descent.[10] NMOSD can correlate with autoimmune diseases such as systemic lupus erythematosus (SLE), celiac disease, Sjögren syndrome, and sarcoidosis.[11] NMOSD attacks have been shown to occur in 20% to 30% of cases after an environmental trigger, such as post-vaccination or infection.[12][13]
Pathophysiology
NMOSD is an inflammatory disease that primarily affects the optic nerve and spinal cord; the brainstem, specifically the area postrema, can also be involved.[14] IgG antibodies against aquaporin-4 are noted in more than 60% to 90% (specificity 90%, sensitivity 70%-90%) of patients with NMOSD.[3][15][16][17][18] Aquaporin-4 is a transmembrane water channel found on the foot processes of astrocytes, and is highly concentrated in certain parts of the central nervous system, such as the optic nerve, the spinal cord, and the area postrema.[19] Circumventricular areas, including periaqueductal grey matter, are rich in AQP4 and are more involved in NMOSD.[18] NMOSD is an autoimmune demyelinating astrocytopathy where the AQP4-IgG mediates perivascular lymphocytic infiltration, leading to axonal loss with a preference for the aforementioned at-risk areas.[20]
Histopathology
Damage to the axons usually precedes demyelination in NMOSD.[17] Histopathological features include activation of microglia or macrophage cells, axonal damage, perivascular deposition of immunoglobulins (especially immunoglobulin M), eosinophilic infiltration, and local activation of the complement system.[21]
History and Physical
Optic neuritis presents with acute onset, painful, monocular vision loss, with a relative afferent pupillary defect in the affected eye.[22] The clinical exam of a patient with acute optic neuritis should always include the visual acuity, pupils, intraocular pressure, and a thorough neuro-ophthalmic exam. A fundus exam will typically reveal optic nerve head elevation on the affected side. Recent literature highlights the less common but more sinister causes of optic neuritis, such as NMOSD or MOGAD; therefore, patients with acute optic neuritis should be specifically asked additional questions during the review of systems. If the patient suffers from intractable hiccups, unexplained nausea or vomiting, or symptomatic narcolepsy, NMOSD may be causing the optic neuritis.[3][23]
NMOSD may alternatively present less classically, and AQP4-IgG can also be present in other disorders (ie, isolated longitudinal extensive transverse myelitis, brainstem clinical manifestations, brainstem encephalitis, monophasic optic neuritis, or recurrent optic neuritis).[11] A patient presenting with neurologic manifestations of brainstem lesions (and intractable hiccups, vomiting, or narcolepsy) should have a complete neurologic exam. Typically, simultaneous bilateral vision loss and paraplegia are noted due to bilateral optic neuritis with transverse myelitis. However, optic neuritis and transverse myelitis may appear weeks apart.[21]
NMOSD has been increasingly recognized as a non-monophasic illness; rather, it follows a relapsing condition with events of disease exacerbation separated by years or decades.[17] Concurrent features of SLE, myasthenia gravis, autoimmune thyroid disease, Sjögren syndrome, and antiphospholipid antibody syndrome may be present.
The core clinical characteristics of NMOSD for adult patients include the following:[3]
- Optic neuritis
- Acute myelitis
- Area postrema syndrome (unexplained hiccups, nausea, or vomiting)
- Acute brainstem syndrome
- Symptomatic narcolepsy or acute diencephalic clinical syndrome with NMOSD-typical diencephalic MRI lesions
- Symptomatic cerebral syndrome with NMOSD-typical brain lesions
Evaluation
NMOSD is evaluated by clinical examination, serologic testing for the AQP4-IgG, and MRI with (and without) gadolinium. Diagnosis is based on core clinical characteristics and supplemented by additional testing. The international consensus diagnostic criteria were established in 2015, separating NMOSD into AQP4-IgG status (with testing, without testing, or unknown) and defining clinical and MRI characteristics of NMOSD.[3] Color vision testing may show a decrease in color vision, and visual field (including Humphrey visual field)[24] evaluation may show a central or centrocecal scotoma. Ancillary testing with optical coherence tomography can aid in the diagnosis and may reveal a thinner peripapillary nerve fiber layer than observed in MS cases.[25]
MRI with and without gadolinium can assist in the evaluation of NMOSD. Characteristic findings in optic neuritis include white matter lesions and optic nerve enhancement (T2-hyperintense lesions or T1-weighted contrast-enhancing images).[3] Other MRI manifestations of NMOSD include acute myelitis, showing longitudinal intramedullary lesions or focal spinal cord atrophic segments.[3] The involvement of bilateral optic nerves, especially of the posterior optic nerve reaching as far as the optic chiasma, is very suggestive of NMOSD. In chronic stages, there is atrophy of the optic nerve and hyperintense signal on T2. The brain lesions in NMOSD may be preferentially located at the location of aquaporin-4 at peri-ependymal regions abutting the ventricles, including confluent periventricular sessile lesions and lesions at the medulla, dorsal pons, medial thalamus, hypothalamus, periaqueductal gray matter, and corpus callosum. The splenium of the corpus callosum may be diffusely involved and swollen. Corpus callosal lesions may have a marbled pattern due to heterogeneous signals from overlapping lesions.[26] Patients with area postrema syndrome demonstrate lesions at the dorsal medulla region or area postrema. Those with acute brainstem syndrome show lesions at the peri-ependymal brainstem. Other brain imaging features include deep white matter lesions (punctate), large white matter lesions without mass effect (especially in pediatric and Black patients), and corticospinal tract lesions (especially in Korean patients).[17][27][28]
When MRI lesions show enhancement in the optic nerves and spinal cord, the main differential diagnosis of NMOSD is MS. It is important to distinguish these clinical entities as the prognosis and management differ considerably, and some MS drugs can worsen NMOSD.[29][30][31] In contrast with MS, NMOSD has some distinct radiologic features that differentiate the condition as a clinical entity. NMOSD shows longitudinal spinal cord lesions extending over 3 or more segments, while MS typically has short, multiple lesions.[27] However, around 14.5% of patients may have short transverse myelitis.[32] Bright spotty lesions in the central gray matter of the spinal cord that are very hyperintense on T2 (more than cerebrospinal fluid) and hypointense on T1 are specific for NMOSD. Acute lesions are usually enhanced with gadolinium contrast in MRI.[32][33]
Treatment / Management
Due to the relative scarcity of NMOSD cases, consensus guidelines are challenging to establish. Most case series recommend a short course of immunosuppressive therapy (usually a corticosteroid), followed by chronic immunosuppressive therapy.[34] Treatment of the acute phase of NMOSD flare usually involves intravenous steroids such as methylprednisolone, often used in high doses (500-100 mg daily) for 5 to 10 days. Other studies advocate plasmapheresis treatment (55 mL/kg) and intravenous immunoglobulin.[35][36]
Chronic immunosuppressive therapy can be accomplished with azathioprine or rituximab as first-line agents.[17] Second-line agents may include mycophenolate or methotrexate.[11] These second-line agents may be advantageous due to their infrequent dosing. Newer biologics are directed against specific immune mediators such as anti-IL-6, anti-complement, or anti-AQP4-IgG.[11] A retrospective multicenter analysis showed reductions in relapse rate with rituximab, mycophenolate, and azathioprine (with prednisone) of 88.2%, 87.4%, and 72.1%, respectively.[37] Differentiating MS from NMOSD is necessary as some drugs used for MS (including natalizumab and beta-interferon) may exacerbate NMOSD.[31][38](B2)
Differential Diagnosis
The differential diagnosis of a patient with optic neuritis typically includes MS, NMOSD, and MOGAD. MS is the most common of these entities and should be ruled out first. The clinician should be especially aware of the other 2 conditions, as they typically portend a less favorable prognosis and require different treatment.
MOGAD is a rare disease associated with antibodies against myelin oligodendrocyte glycoprotein (MOG). MOGAD is usually monophasic (but recurrent optic neuritis may be noted) with a better prognosis than NMOSD. MOGAD has a lower female-to-male ratio or male predominance and is noted more often in childhood. Clinical presentations may include features similar to acute disseminated encephalomyelitis (ADEM), optic neuritis, myelitis, and brainstem encephalitis. MRI features include bilateral anterior involvement of the optic nerve with long length, and the optic chiasma is less involved. The optic nerve head is usually swollen. In the spinal cord, a long segment of the conus or thoracolumbar spinal cord is usually affected. Contrast enhancement is infrequent. In the brain, MRI features include deep gray matter lesions, large lesions similar to ADEM, and fluffy lesions in cerebellar peduncles, pons, and around the fourth ventricle. Lesions of the peri-ependymal areas are rare. Up to 50% of patients with NMOSD or MOGAD can have a normal brain MRI at presentation.[39]
Because NMOSD can also present without ocular symptoms, the presentation and differential of NMOSD broaden significantly when the patient does not have ocular involvement. Still, the primary clinical entity on the differential is MS, but theoretically, any lesion causing MS-like symptoms could be on the differential of NMOSD.
Differences between NMOSD and MS include:[40]
| Features | NMOSD | MS |
| Prevalence | Rare | Common |
| Female | 80% | Around 66%[18] |
| Age at presentation | 40 to 60 years | 20 to 40 years |
| Ethnic predisposition | Asian and Black | White |
| Bilateral optic neuritis | Common | Rare |
| Associated autoimmune diseases | Usually none | May be present, including SLE, myasthenia gravis, autoimmune thyroid disease, Sjögren syndrome, antiphospholipid antibody syndrome |
| Severity of vision loss | Severe with little improvement | Usually moderate visual loss with a good improvement of vision |
| Progressive course | Rare | Common |
| Target tissue | White matter and grey matter | White matter |
| Histopathology | Cavitation or necrosis of tissue is common | Cavitation or necrosis of tissue is rare |
| Imaging of the spinal cord |
Usually longitudinally extensive transverse myelitis (LETM) Patchy, cloud-like enhancement on T1 images with contrast may be present Involvement may extend to the medulla Involvement of the central spinal cord, usually the cervicothoracic segment Usually symptomatic
|
A short segment of the spinal cord is usually involved The Peripheral spinal cord is usually involved May be asymptomatic
|
| Imaging of brainstem |
Involement of dorsal medulla/area postrema The brainstem lesion may be contiguous with the spinal cord lesion |
Clearly demarcated lesions anywhere in the brainstem Any location may be involved, including ventral or dorsal pons |
| Imaging of brain |
Involvement of the corpus callosum is uncommon The thalamus, hypothalamus, and peri-ependymal 3rd ventricle region may be involved |
Dawson fingers (periventricular lesions that are perpendicular to the border of the ventricle and that have a perivenular distribution, more prominent in T2 FLAIR images) The lesions are distributed in time (both new and old lesions) and space (various areas involved [minimum of 2 locations]: typical areas are periventricular, juxtacortical, infratentorial, and the spinal cord) Involvement of corpus callosum is common Involvement of diencephalon is uncommon |
| Imaging of optic nerve |
A long segment of the optic nerve (usually posterior) is usually involved. May be bilateral or unilateral. |
Usually, a short segment of the anterior optic nerve is involved Usually unilateral |
| CSF analysis |
Oligoclonal bands are rare The protein content is higher than MS |
Oligoclonal bands are seen frequently The protein content is lower than MS |
Prognosis
NMOSD has a variable course and prognosis.[23] This condition is either relapsing (80% to 90%) or monophasic.[12] Some patients suffer from a chronic disability as a result of relapsing episodes. Study results have demonstrated that up to 22% of patients fully recover, while 7% do not show any recovery.[23] Individuals with relapsing NMOSD have a worse prognosis. Paraplegia or monoplegia is noted in around 52% of patients with relapsing disease compared to 31% of patients with monophasic disease occurrence.[13] Around 60% of patients with the relapsing disease are blind in 1 or both eyes compared to 22% of patients with the monophasic disease.[13]
Complications
Complications of NMOSD include visual field defects and motor impairment, with potential blindness and irreversible motor deficits.[41] In severe cases, myogenic respiratory failure is the cause of increased mortality.[23]
Deterrence and Patient Education
Patients diagnosed with NMOSD should be educated on potential complications of the disorder, the comorbid-associated autoimmune conditions, and the importance of medication compliance and follow-up.[12] Currently, there are no known preventive strategies for NMOSD.
Enhancing Healthcare Team Outcomes
The neuro-ophthalmologist is often initially involved in diagnosing patients with NMOSD, but patients may also initially present to a primary care provider or neurologist. It is essential to consult with an interprofessional team that includes the ophthalmologist, neurologist, radiologist, and primary care provider to provide adequate care for these patients. The nurse's role is vital as these patients often require hospital admission with rapid induction of intravenous steroids. The pharmacist will be critical in adjusting the appropriate steroid dose for the patient while considering other conditions the patient may have. The radiologist is also key in d appropriately based on MRI results. There aren't any large randomized clinical trials on diagnosing and treating NMOSD; therefore, reliance is primarily on consensus-based expert opinion, and diagnostic criteria are based on historical, case-control studies.[3]
Media
(Click Image to Enlarge)
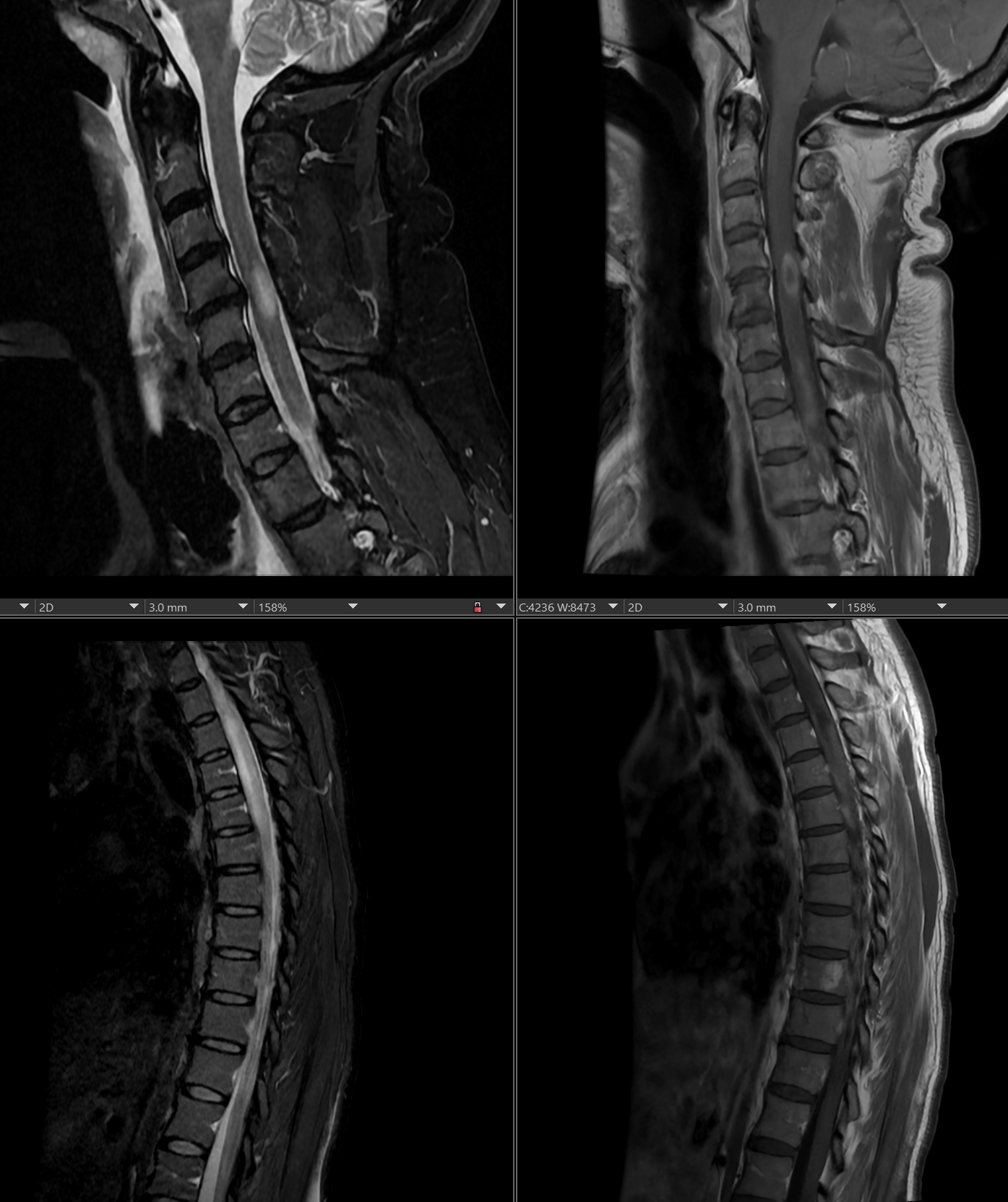
Neuromyelitis Optica. MRI showing inflammation of the spinal column, a common aspect of neuromyelitis optica.
Contributed by Steve Lange, MD
References
Jarius S, Wildemann B. The history of neuromyelitis optica. Journal of neuroinflammation. 2013 Jan 15:10():8. doi: 10.1186/1742-2094-10-8. Epub 2013 Jan 15 [PubMed PMID: 23320783]
Guier CP, Stokkermans TJ. Optic Neuritis. StatPearls. 2023 Jan:(): [PubMed PMID: 32496733]
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, de Seze J, Fujihara K, Greenberg B, Jacob A, Jarius S, Lana-Peixoto M, Levy M, Simon JH, Tenembaum S, Traboulsee AL, Waters P, Wellik KE, Weinshenker BG, International Panel for NMO Diagnosis. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015 Jul 14:85(2):177-89. doi: 10.1212/WNL.0000000000001729. Epub 2015 Jun 19 [PubMed PMID: 26092914]
Level 3 (low-level) evidenceEstrada K, Whelan CW, Zhao F, Bronson P, Handsaker RE, Sun C, Carulli JP, Harris T, Ransohoff RM, McCarroll SA, Day-Williams AG, Greenberg BM, MacArthur DG. A whole-genome sequence study identifies genetic risk factors for neuromyelitis optica. Nature communications. 2018 May 16:9(1):1929. doi: 10.1038/s41467-018-04332-3. Epub 2018 May 16 [PubMed PMID: 29769526]
Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. The Lancet. Neurology. 2012 Jun:11(6):535-44. doi: 10.1016/S1474-4422(12)70133-3. Epub 2012 May 16 [PubMed PMID: 22608667]
Ransohoff RM. Illuminating neuromyelitis optica pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2012 Jan 24:109(4):1001-2. doi: 10.1073/pnas.1119288109. Epub 2012 Jan 23 [PubMed PMID: 22308524]
Level 3 (low-level) evidenceKitley J, Leite MI, Nakashima I, Waters P, McNeillis B, Brown R, Takai Y, Takahashi T, Misu T, Elsone L, Woodhall M, George J, Boggild M, Vincent A, Jacob A, Fujihara K, Palace J. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain : a journal of neurology. 2012 Jun:135(Pt 6):1834-49. doi: 10.1093/brain/aws109. Epub 2012 May 9 [PubMed PMID: 22577216]
Level 2 (mid-level) evidencePandit L, Asgari N, Apiwattanakul M, Palace J, Paul F, Leite MI, Kleiter I, Chitnis T, GJCF International Clinical Consortium & Biorepository for Neuromyelitis Optica. Demographic and clinical features of neuromyelitis optica: A review. Multiple sclerosis (Houndmills, Basingstoke, England). 2015 Jun:21(7):845-53. doi: 10.1177/1352458515572406. Epub 2015 Apr 28 [PubMed PMID: 25921037]
Cabrera-Gómez JA, Kurtzke JF, González-Quevedo A, Lara-Rodríguez R. An epidemiological study of neuromyelitis optica in Cuba. Journal of neurology. 2009 Jan:256(1):35-44. doi: 10.1007/s00415-009-0009-0. Epub 2009 Feb 9 [PubMed PMID: 19224310]
Level 2 (mid-level) evidenceKim SH, Mealy MA, Levy M, Schmidt F, Ruprecht K, Paul F, Ringelstein M, Aktas O, Hartung HP, Asgari N, Tsz-Ching JL, Siritho S, Prayoonwiwat N, Shin HJ, Hyun JW, Han M, Leite MI, Palace J, Kim HJ. Racial differences in neuromyelitis optica spectrum disorder. Neurology. 2018 Nov 27:91(22):e2089-e2099. doi: 10.1212/WNL.0000000000006574. Epub 2018 Oct 26 [PubMed PMID: 30366977]
Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, Borisow N, Kleiter I, Aktas O, Kümpfel T, Neuromyelitis Optica Study Group (NEMOS). Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). Journal of neurology. 2014 Jan:261(1):1-16. doi: 10.1007/s00415-013-7169-7. Epub 2013 Nov 23 [PubMed PMID: 24272588]
Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, Kleiter I, Kleinschnitz C, Berthele A, Brettschneider J, Hellwig K, Hemmer B, Linker RA, Lauda F, Mayer CA, Tumani H, Melms A, Trebst C, Stangel M, Marziniak M, Hoffmann F, Schippling S, Faiss JH, Neuhaus O, Ettrich B, Zentner C, Guthke K, Hofstadt-van Oy U, Reuss R, Pellkofer H, Ziemann U, Kern P, Wandinger KP, Bergh FT, Boettcher T, Langel S, Liebetrau M, Rommer PS, Niehaus S, Münch C, Winkelmann A, Zettl U UK, Metz I, Veauthier C, Sieb JP, Wilke C, Hartung HP, Aktas O, Paul F. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. Journal of neuroinflammation. 2012 Jan 19:9():14. doi: 10.1186/1742-2094-9-14. Epub 2012 Jan 19 [PubMed PMID: 22260418]
Level 2 (mid-level) evidenceWingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology. 1999 Sep 22:53(5):1107-14 [PubMed PMID: 10496275]
Okada K, Kobata M, Naruke S. Neuromyelitis optica spectrum disorder with area postrema syndrome. Neurology. Clinical practice. 2019 Apr:9(2):173-175. doi: 10.1212/CPJ.0000000000000586. Epub [PubMed PMID: 31041136]
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. The Journal of experimental medicine. 2005 Aug 15:202(4):473-7 [PubMed PMID: 16087714]
Level 3 (low-level) evidenceWeinshenker BG, Wingerchuk DM. Neuromyelitis Spectrum Disorders. Mayo Clinic proceedings. 2017 Apr:92(4):663-679. doi: 10.1016/j.mayocp.2016.12.014. Epub [PubMed PMID: 28385199]
Barnett Y, Sutton IJ, Ghadiri M, Masters L, Zivadinov R, Barnett MH. Conventional and advanced imaging in neuromyelitis optica. AJNR. American journal of neuroradiology. 2014 Aug:35(8):1458-66. doi: 10.3174/ajnr.A3592. Epub 2013 Jun 13 [PubMed PMID: 23764723]
Sarbu N, Shih RY, Jones RV, Horkayne-Szakaly I, Oleaga L, Smirniotopoulos JG. White Matter Diseases with Radiologic-Pathologic Correlation. Radiographics : a review publication of the Radiological Society of North America, Inc. 2016 Sep-Oct:36(5):1426-47. doi: 10.1148/rg.2016160031. Epub [PubMed PMID: 27618323]
Romeo AR, Segal BM. Treatment of neuromyelitis optica spectrum disorders. Current opinion in rheumatology. 2019 May:31(3):250-255. doi: 10.1097/BOR.0000000000000603. Epub [PubMed PMID: 30920972]
Level 3 (low-level) evidenceWingerchuk DM. Evidence for humoral autoimmunity in neuromyelitis optica. Neurological research. 2006 Apr:28(3):348-53 [PubMed PMID: 16687064]
Level 3 (low-level) evidenceGold R, Linington C. Devic's disease: bridging the gap between laboratory and clinic. Brain : a journal of neurology. 2002 Jul:125(Pt 7):1425-7 [PubMed PMID: 12076994]
Level 3 (low-level) evidenceSimakurthy S, Tripathy K. Marcus Gunn Pupil. StatPearls. 2025 Jan:(): [PubMed PMID: 32491607]
Patterson SL, Goglin SE. Neuromyelitis Optica. Rheumatic diseases clinics of North America. 2017 Nov:43(4):579-591. doi: 10.1016/j.rdc.2017.06.007. Epub 2017 Aug 31 [PubMed PMID: 29061244]
Ruia S, Tripathy K. Humphrey Visual Field. StatPearls. 2025 Jan:(): [PubMed PMID: 36256759]
de Seze J, Blanc F, Jeanjean L, Zéphir H, Labauge P, Bouyon M, Ballonzoli L, Castelnovo G, Fleury M, Defoort S, Vermersch P, Speeg C. Optical coherence tomography in neuromyelitis optica. Archives of neurology. 2008 Jul:65(7):920-3. doi: 10.1001/archneur.65.7.920. Epub [PubMed PMID: 18625858]
Level 2 (mid-level) evidenceNakamura M, Misu T, Fujihara K, Miyazawa I, Nakashima I, Takahashi T, Watanabe S, Itoyama Y. Occurrence of acute large and edematous callosal lesions in neuromyelitis optica. Multiple sclerosis (Houndmills, Basingstoke, England). 2009 Jun:15(6):695-700. doi: 10.1177/1352458509103301. Epub 2009 May 12 [PubMed PMID: 19435750]
Level 3 (low-level) evidenceKim HJ, Paul F, Lana-Peixoto MA, Tenembaum S, Asgari N, Palace J, Klawiter EC, Sato DK, de Seze J, Wuerfel J, Banwell BL, Villoslada P, Saiz A, Fujihara K, Kim SH, Guthy-Jackson Charitable Foundation NMO International Clinical Consortium & Biorepository. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. 2015 Mar 17:84(11):1165-73. doi: 10.1212/WNL.0000000000001367. Epub 2015 Feb 18 [PubMed PMID: 25695963]
Level 3 (low-level) evidenceKim W, Lee JE, Kim SH, Huh SY, Hyun JW, Jeong IH, Park MS, Cho JY, Lee SH, Lee KS, Kim HJ. Cerebral Cortex Involvement in Neuromyelitis Optica Spectrum Disorder. Journal of clinical neurology (Seoul, Korea). 2016 Apr:12(2):188-93. doi: 10.3988/jcn.2016.12.2.188. Epub 2016 Jan 28 [PubMed PMID: 26833983]
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Multiple sclerosis (Houndmills, Basingstoke, England). 2012 Oct:18(10):1480-3 [PubMed PMID: 22354738]
Level 2 (mid-level) evidenceMin JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Multiple sclerosis (Houndmills, Basingstoke, England). 2012 Jan:18(1):113-5. doi: 10.1177/1352458511431973. Epub 2011 Dec 6 [PubMed PMID: 22146605]
Level 3 (low-level) evidenceJacob A, Hutchinson M, Elsone L, Kelly S, Ali R, Saukans I, Tubridy N, Boggild M. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012 Sep 4:79(10):1065-6. doi: 10.1212/WNL.0b013e31826845fe. Epub 2012 Aug 22 [PubMed PMID: 22914835]
Level 3 (low-level) evidenceDutra BG, da Rocha AJ, Nunes RH, Maia ACM Júnior. Neuromyelitis Optica Spectrum Disorders: Spectrum of MR Imaging Findings and Their Differential Diagnosis. Radiographics : a review publication of the Radiological Society of North America, Inc. 2018 Jan-Feb:38(1):169-193. doi: 10.1148/rg.2018170141. Epub [PubMed PMID: 29320331]
Zalewski NL, Morris PP, Weinshenker BG, Lucchinetti CF, Guo Y, Pittock SJ, Krecke KN, Kaufmann TJ, Wingerchuk DM, Kumar N, Flanagan EP. Ring-enhancing spinal cord lesions in neuromyelitis optica spectrum disorders. Journal of neurology, neurosurgery, and psychiatry. 2017 Mar:88(3):218-225. doi: 10.1136/jnnp-2016-314738. Epub 2016 Dec 2 [PubMed PMID: 27913626]
Pearce JM. Neuromyelitis optica. Spinal cord. 2005 Nov:43(11):631-4 [PubMed PMID: 15968305]
Gómez-Figueroa E, Alvarado-Bolaños A, García-Estrada C, Zabala-Ángeles I, Sánchez-Rosales N, Bribiesca-Contreras E, García-Alvarez G, Montes-Pérez Y, Ramos-Vega E, Casallas-Vanegas A, Carrillo-Loza K, Corona-Vázquez T, Rivas-Alonso V, Flores-Rivera J. Clinical experience of plasmapheresis for neuromyelitis optica patients in Mexico. Multiple sclerosis and related disorders. 2021 Jul:52():103022. doi: 10.1016/j.msard.2021.103022. Epub 2021 May 15 [PubMed PMID: 34034213]
Li X, Tian DC, Fan M, Xiu Y, Wang X, Li T, Jia D, Xu W, Song T, Shi FD, Zhang X. Intravenous immunoglobulin for acute attacks in neuromyelitis optica spectrum disorders (NMOSD). Multiple sclerosis and related disorders. 2020 Sep:44():102325. doi: 10.1016/j.msard.2020.102325. Epub 2020 Jun 26 [PubMed PMID: 32653803]
Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA neurology. 2014 Mar:71(3):324-30. doi: 10.1001/jamaneurol.2013.5699. Epub [PubMed PMID: 24445513]
Level 2 (mid-level) evidenceAgasing A, Quinn JL, Kumar G, Axtell RC. Interferon-β Intensifies Interleukin-23-Driven Pathogenicity of T Helper Cells in Neuroinflammatory Disease. Cells. 2021 Aug 20:10(8):. doi: 10.3390/cells10082139. Epub 2021 Aug 20 [PubMed PMID: 34440908]
Hegen H, Reindl M. Recent developments in MOG-IgG associated neurological disorders. Therapeutic advances in neurological disorders. 2020:13():1756286420945135. doi: 10.1177/1756286420945135. Epub 2020 Jul 31 [PubMed PMID: 33029200]
Level 3 (low-level) evidenceHuda S, Whittam D, Bhojak M, Chamberlain J, Noonan C, Jacob A. Neuromyelitis optica spectrum disorders. Clinical medicine (London, England). 2019 Mar:19(2):169-176. doi: 10.7861/clinmedicine.19-2-169. Epub [PubMed PMID: 30872305]
Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, Du Pasquier RA, Polman CH, Sorensen PS, Hemmer B. EFNS guidelines on diagnosis and management of neuromyelitis optica. European journal of neurology. 2010 Aug:17(8):1019-32. doi: 10.1111/j.1468-1331.2010.03066.x. Epub 2010 Jun 7 [PubMed PMID: 20528913]