Introduction
Urbach and Wiethe first described lipoid Proteinosis (LP) (hyalinosis cutis et mucosae) in 1929.[1] LP is a rare genodermatosis characterized by the deposition of hyaline-like material in various tissues and organs. LP is associated with a variety of characteristic cutaneous and mucosal findings and several neurologic, psychiatric, and gastrointestinal manifestations.[2][3][4][5][6]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
LP is caused by a homozygous or compound heterozygous loss-of-function mutation in the extracellular matrix protein 1 (ECM1) gene located on chromosome 1q21.[7][8][9] Most ECM1 mutations are frameshift or nonsense mutations in exons 6 or 7, resulting in a truncated ECM1.[8] The ECM1 gene encodes glycoproteins essential for basement membrane and extracellular matrix structure integrity, skin adhesion, and protein-protein interactions.[9][10] Since there are various ECM1 splice variants and mutations, many LP genotypes and phenotypes are possible.[8][10]
LP is inherited in an autosomal recessive pattern. Patients often have affected family members, and some are children of consanguineous parents.
Epidemiology
LP occurs worldwide but is rare, with only about 400 cases reported in the medical literature.[10][11][12] Males and females are affected equally.[9] The Namaqualand region in South Africa has the largest LP patients sharing a common mutation, suggesting a founder effect.[12] There is a higher incidence of LP in countries where consanguinity is more common.[13][14]
Pathophysiology
ECM1 is a glycoprotein that plays various extracutaneous roles, including regulation of endochondral bone and cartilage formation, endothelial cell proliferation and angiogenesis, and dendritic cell differentiation.[15] High levels of ECM1 expression may be associated with certain malignancies.[16][17]
In the skin, ECM1 interacts with basement membrane-specific heparan sulfate proteoglycan core protein (HSPG), also known as perlecan, and other proteins important for ECM structure and tissue remodeling like matrix metalloproteinase-9, fibulin-3, and laminin 332.[10][18][19] Certain ECM1 splice variants are expressed within basal keratinocytes and suprabasal cells, indicating a possible role in keratinocyte differentiation.[20] ECM1 expression is reduced in individuals with chronic ultraviolet light exposure, suggesting an additional role in the cutaneous stress response.[21] Ultrastructural analysis of erosive lesions in a child revealed “free-floating” desmosomes as well as connection loss between some desmosomes and the keratin filament network. This points to a possible role for ECM1 in the attachment between keratin filaments and the desmosome.[22]
Mutations within the ECM1 gene lead to compromised protein-protein interactions and disrupted tissue homeostasis.[8][9][10] Hyaline-like accumulation within different tissues explains many classic clinical findings like hoarseness, moniliform blepharosis, and seizures.
Histopathology
Histologically, LP is characterized by hyaline-like periodic acid-Schiff (PAS)-positive, diastase-resistant deposits within the papillary dermis and at the dermal-epidermal junction (DEJ).[23][24] The basement membrane surrounding blood vessels at the DEJ is reduplicated and structurally disrupted.[25][26] Other blood vessel abnormalities include marked dilation and absence of several groups of vessels (including the dermal papillae capillary loop network, the subcutaneous plexus, and the transverse connecting vessels).[24] Additional histopathologic findings include sweat gland hyalinization and hyperkeratosis.
Immunolabeling can be a useful diagnostic tool, especially early in the disease course. Reduced ECM1 protein in conjunction with characteristic clinical findings should heighten suspicion for LP.[27] Congo-red staining can help differentiate LP from amyloidosis, which is often on the differential diagnosis.
History and Physical
In almost all affected individuals, the earliest manifestation is a weak or hoarse cry in early life due to hyaline-like deposits on the vocal cords.[10][28][29] Hoarseness and severe dysphonia (“husky” voice) can develop and persist throughout life.[30] Deposits within the larynx can occasionally obstruct the airway.[31]
Cutaneous abnormalities also present early in the disease course, usually beginning with vesicles or bullae and hemorrhagic crusts on sites of friction or trauma, most notably the face and around the mouth.[13]These lesions heal with prominent pox-like or acneiform scarring (Figure A).[32] Over time, the skin can appear thick and waxy with a yellowish hue.[29] Moniliform blepharosis, “beaded” papules along the eyelid margin, is a pathognomonic finding identified in about 50% of patients (Figure B).[33] Other cutaneous manifestations include papules, nodules, plaques on the face and lips, and verrucous and hyperkeratotic lesions at sites of trauma or friction.[29] Early clinical clues, namely cutaneous buttocks lesions, may predict a more severe clinical course with neurologic involvement.[34] There have been infrequent reports of patchy or diffuse alopecia at lesional sites.
Other notable features of LP reflect the infiltrating hyaline-like material throughout the mouth and upper aerodigestive tract. Cobblestone appearance of the oral mucosa, yellow lip nodules, lip eversion, and vegetative lesions and fissures at the lateral commissures can be seen in patients with extensive disease.[6][29][31]
As a result of deposits obstructing Stenson’s duct and secondary xerostomia, patients with LP are prone to frequent upper respiratory tract infections, recurrent parotitis, and dental caries. The submandibular gland can become similarly obstructed and inflamed.[10] The tongue is often “woody” to palpation, with absent dorsal papillae, a shortened and thickened frenulum, and restricted protrusion.[6][35]
Neurologic manifestations of LP can be severe, including epilepsy, dystonia, progressive neuropsychiatric disorders (memory loss and other cognitive impairment, behavioral changes, hallucination, schizophreniform illness), and spontaneous central nervous system (CNS) hemorrhage.[3][36] Hyaline-like material can be deposited throughout the CNS with or without consequent symptomatology.[37] A post-mortem neuropathologic analysis showed calcium accumulation with evidence of adjacent gliosis, vessel calcification, and occlusions with perivascular infarctions, and demyelination.[37]
Bilateral, symmetric comma-shaped calcification of the amygdala is a pathognomonic radiologic finding. Calcification has also been reported in the hippocampus, basal ganglia, parahippocampal gyrus, uncinate gyrus, striatum, pineal gland, and perirhinal and parietal cortices.[37][38][39] Calcifications develop slowly and are therefore seen more frequently in patients greater than age 10 and in those with longer disease duration.[37][40]
One study of 10 patients with LP radiographically identified six with amygdaloid complex calcification and degeneration and three with decreased perfusion to the medial temporal lobe, suspicious for underlying calcification. Though these nine patients did not differ from healthy controls for cognitive tasks (memory, attention, executive function), they differed from recognizing facial expression, emotional processing, and odor-associative learning and recognition.[39] In an analysis of 7 patients with intracranial calcifications, four had epilepsy with onset in childhood or young adulthood.[36]
Focal seizures were the most common subtype, as demonstrated by video electroencephalography. Patients with certain mutations tended to be resistant to anti-epileptic therapy while others attained seizure-control with levetiracetam and clobazam.[36]
Finally, there have been rare reports of hyaline-like deposits throughout the digestive tract, some of which regressed with time.[4] One patient presented with acute gastrointestinal hemorrhage as a result of deposits within the small bowel.[4][5]
Evaluation
Because of its autosomal recessive inheritance pattern, patients with LP should be asked about family members with similar symptoms and consanguinity. Synthesis of family history, clinical exam, and histopathologic findings often raise suspicion for LP, though definitive diagnosis can only be made upon identifying an ECM1 mutation.
An otolaryngologist should evaluate patients to assess airway involvement and identify any degree of obstruction. Similarly, patients with neurologic or neuropsychiatric symptoms merit evaluation and follow-up with a neurologist and psychiatrist. Neuroimaging may become necessary to identify involved brain regions.
Other evaluations will depend on disease manifestations but should include frequent dental, dermatologic, and ophthalmic evaluation.
Treatment / Management
There are been no randomized trials to establish a standard of care for LP. Treatment plans should be individualized based on manifestations of disease and desire for improved cosmetic outcomes.
Systemic retinoids such as acitretin or etretinate may be effective in treating cutaneous and laryngeal lesions by decreasing the amount of hyaline-like deposits within the dermis.[41] Case reports describing long-term acitretin therapy (0.5mg/kg per day) have noted healing of ulcerated and vesicobullous lesions decrease in tongue size with an improved protrusion,[42] diminished hoarseness and softening of hyperkeratotic verrucous papules and nodules.[43] A few patients who saw improved voice quality had limited, if any, cutaneous improvement.[44][45](B3)
There is conflicting evidence about the efficacy of long-term oral dimethyl sulfoxide (DMSO) for the treatment of cutaneous lesions and hoarseness.[46][47] One case report noted clinical improvement with the chelating agent D-penicillamine 600mg/day.[48](B3)
For patients with vocal cord involvement and impaired phonation, microlaryngoscopy and carbon dioxide (CO) laser procedures can be quite successful.[49][50] However, there is a risk of postoperative granulation tissue formation, fibrosis, and the need for follow-up interventions. For this reason, some argue that surgical intervention is only indicated with a compromised airway. (B3)
To minimize the appearance of scars and moniliform blepharosis, cosmetic procedures such as CO laser, dermabrasion, blepharoplasty, and erbium-doped yttrium aluminum garnet ablative laser have been employed.[51][52][53](B3)
Management of neurologic and psychiatric manifestations of LP should be tailored to each individual. Certain anti-epileptic medications can minimize seizure frequency and severity. Anti-psychotic medication can help manage behavioral changes and psychosis.
Differential Diagnosis
Differential diagnoses may differ between patients. Possible differential diagnoses include:
Cutaneous lesions:
- Herpes simplex infection
- Impetigo
- Epidermolysis bullosa
- Erythropoietic protoporphyria
- Lichen myxedematosus
- Systemic amyloidosis
- Nodular localized cutaneous amyloidosis
- Colloid milium
- Hyalinosis
- Xanthomas
- Scleromyxedema
- Leprosy
- Hydroa vacciniforme
- Incontinentia pigmenti
Hoarseness:
- Laryngitis
- Laryngotracheitis
- Laryngotracheobronchitis
- Vocal cord nodules
- Gastroesophageal reflux disease
- Vocal cord polyps
- Laryngeal hemangiomas
- Laryngeal cysts
- Vocal cord paralysis
- Vocal cord hypertrophy
Enlarged tongue:
- Systemic amyloidosis
- Hypothyroidism (congenital)
- Myxedema
- Acromegaly
Mesial temporal lobe calcification:
- Fahr’s disease
- Calcified glioma
- Raine syndrome
- Healed herpes encephalitis
- Dystonia
Prognosis
LP typically runs a benign but progressive course that is compatible with a normal lifespan. Respiratory obstruction and central nervous system involvement, however, can be life-threatening or impart significant morbidity.
Complications
LP can cause a wide variety of clinical manifestations due to hyaline deposition in the skin, mucosa, and various organs. Of the possible clinical complications, airway obstruction, seizures, spontaneous CNS hemorrhage are the most severe. Hyaline deposition within the small bowel has also been linked to cases of gastrointestinal bleeding.[4][5]
There can also be complications as a result of surgical intervention on the vocal cords or larynx. These include bleeding, infection, and vocal cord scarring leading to a change in voice quality and a possible need for repeat intervention.
Finally, certain patients may be taking systemic medication to manage their disease, such as anti-psychotic, anti-epileptic, and dopamine-modifying agents. These medications can have a variety of drug-specific side effects.
Consultations
A multidisciplinary approach is recommended for patients with LP. Counseling may involve a dermatologist, otolaryngologist, neurologist, psychiatrist, dentist, and geneticist depending on manifestations of the disease. Like other autosomal recessive conditions, carrier testing and reproductive counseling should be offered to patients and their family members.
Enhancing Healthcare Team Outcomes
LP is a rare genetic disease that requires interprofessional collaboration for optimal patient care. Patients with suspected LP should undergo a comprehensive workup with several specialists, including a dermatologist, neurologist, psychiatrist, otolaryngologist, dentist, and geneticist. It is important to assess disease burden and to evaluate for any possibly life-threatening manifestations. Patients’ symptoms and quality of life should be assessed frequently. Additionally, genetic counseling may offer patients a better understanding of their condition and its mode of inheritance.
Media
(Click Image to Enlarge)
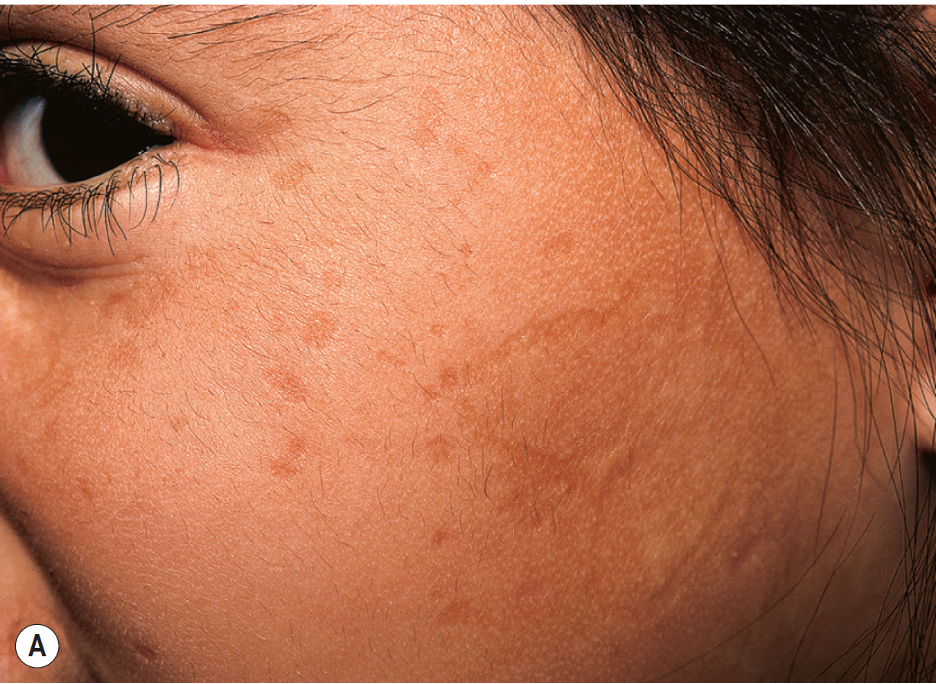
Figure A: Scarring on the face of a child following vesicle formation and hemorrhagic crusting. Figures A and B were published in Hurwitz Clinical Pediatric Dermatology, Fifth Edition, Amy S. Paller MD and Anthony J. Mancini MD, Chapter 6-Hereditary Disorders of the Dermis, Pages 119-135, Copyright Elsevier (2016).
(Click Image to Enlarge)
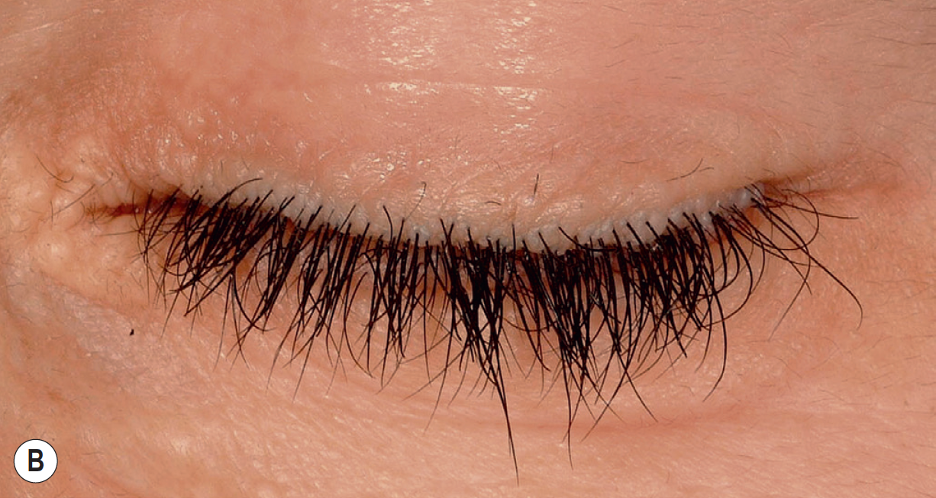
Figure B: Moniliform blepharosis, or “beaded” papulation along the eyelid margin, is a feature seen in about 50% of patients with LP. Figures A and B were published in Hurwitz Clinical Pediatric Dermatology, Fifth Edition, Amy S. Paller MD and Anthony J. Mancini MD, Chapter 6-Hereditary Disorders of the Dermis, Pages 119-135, Copyright Elsevier (2016).
References
KATZENELLENBOGEN I,UNGAR H, Lipoid proteinosis; reinvestigation of a case previously reported by Urbach and Wiethe in 1929. Dermatologica. 1957 Jul; [PubMed PMID: 13461538]
Level 3 (low-level) evidenceTeive HA,Ruschel E,Munhoz RP, Spontaneous intracerebral hemorrhage in Urbach-Wiethe disease. Neurology. 2013 Apr 30; [PubMed PMID: 23628933]
Level 3 (low-level) evidenceMcgrath JA, Lipoid proteinosis. Handbook of clinical neurology. 2015; [PubMed PMID: 26564090]
Custódio Lima J,Nagasako CK,Montes CG,Barcelos IH,de Carvalho RB,Mesquita MA, Gastrointestinal involvement in lipoid proteinosis: a ten-year follow-up of a brazilian female patient. Case reports in medicine. 2014; [PubMed PMID: 25045357]
Level 3 (low-level) evidenceCaccamo D,Jaen A,Telenta M,Varela E,Tiscornia O, Lipoid proteinosis of the small bowel. Archives of pathology [PubMed PMID: 7514865]
Level 3 (low-level) evidenceFrenkel B,Vered M,Taicher S,Yarom N, Lipoid proteinosis unveiled by oral mucosal lesions: a comprehensive analysis of 137 cases. Clinical oral investigations. 2017 Sep; [PubMed PMID: 27900487]
Level 3 (low-level) evidenceNasir M,Latif A,Ajmal M,Qamar R,Naeem M,Hameed A, Molecular analysis of lipoid proteinosis: identification of a novel nonsense mutation in the ECM1 gene in a Pakistani family. Diagnostic pathology. 2011 Jul 26; [PubMed PMID: 21791056]
Hamada T,Wessagowit V,South AP,Ashton GH,Chan I,Oyama N,Siriwattana A,Jewhasuchin P,Charuwichitratana S,Thappa DM,Jeevankumar B,Lenane P,Krafchik B,Kulthanan K,Shimizu H,Kaya TI,Erdal ME,Paradisi M,Paller AS,Seishima M,Hashimoto T,McGrath JA, Extracellular matrix protein 1 gene (ECM1) mutations in lipoid proteinosis and genotype-phenotype correlation. The Journal of investigative dermatology. 2003 Mar; [PubMed PMID: 12603844]
Level 3 (low-level) evidenceHamada T,McLean WH,Ramsay M,Ashton GH,Nanda A,Jenkins T,Edelstein I,South AP,Bleck O,Wessagowit V,Mallipeddi R,Orchard GE,Wan H,Dopping-Hepenstal PJ,Mellerio JE,Whittock NV,Munro CS,van Steensel MA,Steijlen PM,Ni J,Zhang L,Hashimoto T,Eady RA,McGrath JA, Lipoid proteinosis maps to 1q21 and is caused by mutations in the extracellular matrix protein 1 gene (ECM1). Human molecular genetics. 2002 Apr 1; [PubMed PMID: 11929856]
Chan I,Liu L,Hamada T,Sethuraman G,McGrath JA, The molecular basis of lipoid proteinosis: mutations in extracellular matrix protein 1. Experimental dermatology. 2007 Nov; [PubMed PMID: 17927570]
Di Giandomenico S,Masi R,Cassandrini D,El-Hachem M,De Vito R,Bruno C,Santorelli FM, Lipoid proteinosis: case report and review of the literature. Acta otorhinolaryngologica Italica : organo ufficiale della Societa italiana di otorinolaringologia e chirurgia cervico-facciale. 2006 Jun; [PubMed PMID: 17063986]
Level 3 (low-level) evidenceVan Hougenhouck-Tulleken W,Chan I,Hamada T,Thornton H,Jenkins T,McLean WH,McGrath JA,Ramsay M, Clinical and molecular characterization of lipoid proteinosis in Namaqualand, South Africa. The British journal of dermatology. 2004 Aug; [PubMed PMID: 15327549]
Level 2 (mid-level) evidenceNanda A,Alsaleh QA,Al-Sabah H,Ali AM,Anim JT, Lipoid proteinosis: report of four siblings and brief review of the literature. Pediatric dermatology. 2001 Jan-Feb; [PubMed PMID: 11207965]
Level 3 (low-level) evidenceYoussefian L,Vahidnezhad H,Daneshpazhooh M,Abdollahzadeh S,Talari H,Khoshnevisan A,Chams-Davatchi C,Mobasher R,Li Q,Uitto J,Akhondzadeh S,Tabrizi M, Lipoid proteinosis: phenotypic heterogeneity in Iranian families with c.507delT mutation in ECM1. Experimental dermatology. 2015 Mar; [PubMed PMID: 25529926]
Level 3 (low-level) evidenceLe Naour F,Hohenkirk L,Grolleau A,Misek DE,Lescure P,Geiger JD,Hanash S,Beretta L, Profiling changes in gene expression during differentiation and maturation of monocyte-derived dendritic cells using both oligonucleotide microarrays and proteomics. The Journal of biological chemistry. 2001 May 25; [PubMed PMID: 11279020]
Han Z,Ni J,Smits P,Underhill CB,Xie B,Chen Y,Liu N,Tylzanowski P,Parmelee D,Feng P,Ding I,Gao F,Gentz R,Huylebroeck D,Merregaert J,Zhang L, Extracellular matrix protein 1 (ECM1) has angiogenic properties and is expressed by breast tumor cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001 Apr; [PubMed PMID: 11292659]
Rickman DS,Bobek MP,Misek DE,Kuick R,Blaivas M,Kurnit DM,Taylor J,Hanash SM, Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer research. 2001 Sep 15; [PubMed PMID: 11559565]
Sercu S,Lambeir AM,Steenackers E,El Ghalbzouri A,Geentjens K,Sasaki T,Oyama N,Merregaert J, ECM1 interacts with fibulin-3 and the beta 3 chain of laminin 332 through its serum albumin subdomain-like 2 domain. Matrix biology : journal of the International Society for Matrix Biology. 2009 Apr; [PubMed PMID: 19275936]
Fujimoto N,Terlizzi J,Aho S,Brittingham R,Fertala A,Oyama N,McGrath JA,Uitto J, Extracellular matrix protein 1 inhibits the activity of matrix metalloproteinase 9 through high-affinity protein/protein interactions. Experimental dermatology. 2006 Apr; [PubMed PMID: 16512877]
Smits P,Poumay Y,Karperien M,Tylzanowski P,Wauters J,Huylebroeck D,Ponec M,Merregaert J, Differentiation-dependent alternative splicing and expression of the extracellular matrix protein 1 gene in human keratinocytes. The Journal of investigative dermatology. 2000 Apr; [PubMed PMID: 10733679]
Sander CS,Sercu S,Ziemer M,Hipler UC,Elsner P,Thiele JJ,Merregaert J, Expression of extracellular matrix protein 1 (ECM1) in human skin is decreased by age and increased upon ultraviolet exposure. The British journal of dermatology. 2006 Feb; [PubMed PMID: 16433788]
Olsen DR,Chu ML,Uitto J, Expression of basement membrane zone genes coding for type IV procollagen and laminin by human skin fibroblasts in vitro: elevated alpha 1 (IV) collagen mRNA levels in lipoid proteinosis. The Journal of investigative dermatology. 1988 May; [PubMed PMID: 3361143]
Level 3 (low-level) evidenceKowalewski C,Kozłowska A,Chan I,Górska M,Woźniak K,Jabłońska S,McGrath JA, Three-dimensional imaging reveals major changes in skin microvasculature in lipoid proteinosis and lichen sclerosus. Journal of dermatological science. 2005 Jun; [PubMed PMID: 15927815]
Level 3 (low-level) evidenceFabrizi G,Porfiri B,Borgioli M,Serri F, Urbach-Wiethe disease. Light and electron microscopic study. Journal of cutaneous pathology. 1980 Feb; [PubMed PMID: 7358884]
Level 3 (low-level) evidenceMoy LS,Moy RL,Matsuoka LY,Ohta A,Uitto J, Lipoid proteinosis: ultrastructural and biochemical studies. Journal of the American Academy of Dermatology. 1987 Jun; [PubMed PMID: 2439554]
Level 3 (low-level) evidenceChan I,South AP,McGrath JA,Oyama N,Bhogal BS,Black MM,Hamada T, Rapid diagnosis of lipoid proteinosis using an anti-extracellular matrix protein 1 (ECM1) antibody. Journal of dermatological science. 2004 Aug; [PubMed PMID: 15265527]
Level 3 (low-level) evidenceSriwastava S,Desai A,Yuliati A,Watson CR,Sivaswamy L, A Child With a Hoarse Cry and Intracranial Calcification. Pediatric neurology. 2018 Oct; [PubMed PMID: 30501889]
Hamada T, Lipoid proteinosis. Clinical and experimental dermatology. 2002 Nov; [PubMed PMID: 12472532]
Savage MM,Crockett DM,McCabe BF, Lipoid proteinosis of the larynx: a cause of voice change in the infant and young child. International journal of pediatric otorhinolaryngology. 1988 Feb; [PubMed PMID: 3372140]
Level 3 (low-level) evidenceAlly M,Kinshuck AJ,Sandison A,Sandhu GS, The management of laryngeal lipoid proteinosis. The Journal of laryngology and otology. 2018 Oct; [PubMed PMID: 30099970]
Nayak S,Acharjya B, Lipoid proteinosis in a six-year-old child. Indian dermatology online journal. 2012 Jan; [PubMed PMID: 23130256]
Level 3 (low-level) evidenceBelliveau MJ,Alkhotani A,Ali A, Moniliform Blepharosis of Lipoid Proteinosis. JAMA ophthalmology. 2015 Jul; [PubMed PMID: 26158437]
Hamie L,Knio Z,Abbas O,Akel R,Bardawil T,Kibbi AG,Kurban M, Clinical clues early in the lives of individuals with lipoid proteinosis can determine the course of the disease. Clinical and experimental dermatology. 2017 Jun; [PubMed PMID: 28244239]
Bozdağ KE,Gül Y,Karaman A, Lipoid proteinosis. International journal of dermatology. 2000 Mar; [PubMed PMID: 10759961]
Level 3 (low-level) evidenceOguz Akarsu E,Dinçsoy Bir F,Baykal C,Taşdemir V,Kara B,Bebek N,Gürses C,Uyguner O,Baykan B, The Characteristics and Long-Term Course of Epilepsy in Lipoid Proteinosis: A Spectrum From Mild to Severe Seizures in Relation to ECM1 Mutations. Clinical EEG and neuroscience. 2018 May; [PubMed PMID: 28434238]
Appenzeller S,Chaloult E,Velho P,de Souza EM,Araújo VZ,Cendes F,Li LM, Amygdalae calcifications associated with disease duration in lipoid proteinosis. Journal of neuroimaging : official journal of the American Society of Neuroimaging. 2006 Apr; [PubMed PMID: 16629738]
Level 3 (low-level) evidenceChandrasekaran S,Nanjundan M,Natarajan S,Ramadhas K, Radiologic presentation of lipoid proteinosis with symmetrical medial temporal lobe calcifications. Radiology case reports. 2015; [PubMed PMID: 27398129]
Level 3 (low-level) evidenceSiebert M,Markowitsch HJ,Bartel P, Amygdala, affect and cognition: evidence from 10 patients with Urbach-Wiethe disease. Brain : a journal of neurology. 2003 Dec; [PubMed PMID: 12937075]
Dertlioğlu SB,Çalık M,Çiçek D, Demographic, clinical, and radiologic signs and treatment responses of lipoid proteinosis patients: a 10-case series from Şanlıurfa. International journal of dermatology. 2014 Apr; [PubMed PMID: 24320796]
Level 2 (mid-level) evidenceFritsch PO, Retinoids in psoriasis and disorders of keratinization. Journal of the American Academy of Dermatology. 1992 Dec; [PubMed PMID: 1460124]
Bakry OA,Samaka RM,Houla NS,Basha MA, Two Egyptian cases of lipoid proteinosis successfully treated with acitretin. Journal of dermatological case reports. 2014 Mar 31; [PubMed PMID: 24748909]
Level 3 (low-level) evidenceAkoglu G,Karaduman A,Ergin S,Erkin G,Gokoz O,Unal OF,Hamada T, Clinical and histopathological response to acitretin therapy in lipoid proteinosis. The Journal of dermatological treatment. 2011 Jun; [PubMed PMID: 20666665]
Level 3 (low-level) evidenceToosi S,Ehsani AH, Treatment of lipoid proteinosis with acitretin: a case report. Journal of the European Academy of Dermatology and Venereology : JEADV. 2009 Apr; [PubMed PMID: 18808438]
Level 3 (low-level) evidenceGündüz O,Sahiner N,Atasoy P,Senyücel C, Acitretin treatment for lipoid proteinosis. Case reports in dermatological medicine. 2012; [PubMed PMID: 23259080]
Level 3 (low-level) evidenceWong CK,Lin CS, Remarkable response of lipoid proteinosis to oral dimethyl sulphoxide. The British journal of dermatology. 1988 Oct; [PubMed PMID: 3191019]
Level 3 (low-level) evidenceOzkaya-Bayazit E,Ozarmağan G,Baykal C,Uluğ T, [Oral DMSO therapy in 3 patients with lipoidproteinosis. Results of long-term therapy]. Der Hautarzt; Zeitschrift fur Dermatologie, Venerologie, und verwandte Gebiete. 1997 Jul; [PubMed PMID: 9333627]
Level 3 (low-level) evidenceKaya TI,Kokturk A,Tursen U,Ikizoglu G,Polat A, D-penicillamine treatment for lipoid proteinosis. Pediatric dermatology. 2002 Jul-Aug; [PubMed PMID: 12220287]
Level 3 (low-level) evidenceXu W,Wang L,Zhang L,Liu HG,Zan F,Hu R,Liu WB,Han DM, [Manifestation and treatment of lipoid proteinosis in larynx]. Zhonghua er bi yan hou tou jing wai ke za zhi = Chinese journal of otorhinolaryngology head and neck surgery. 2010 Apr; [PubMed PMID: 20627049]
Harper JI,Duguid KP,Staughton RC,Moffat DA, Oropharyngeal and laryngeal lesions in lipoid proteinosis. The Journal of laryngology and otology. 1983 Sep; [PubMed PMID: 6886551]
Level 3 (low-level) evidenceRosenthal G,Lifshitz T,Monos T,Kachco L,Argov S, Carbon dioxide laser treatment for lipoid proteinosis (Urbach-Wiethe syndrome) involving the eyelids. The British journal of ophthalmology. 1997 Mar; [PubMed PMID: 9135394]
Level 3 (low-level) evidenceBuchan NG,Kemble JV, Successful surgical treatment of lipoid proteinosis. The British journal of dermatology. 1974 May; [PubMed PMID: 4833793]
Çalıskan E,Açıkgöz G,Tunca M,Koç E,Arca E,Akar A, Treatment of lipoid proteinosis with ablative Er:YAG laser resurfacing. Dermatologic therapy. 2015 Sep-Oct; [PubMed PMID: 26031844]