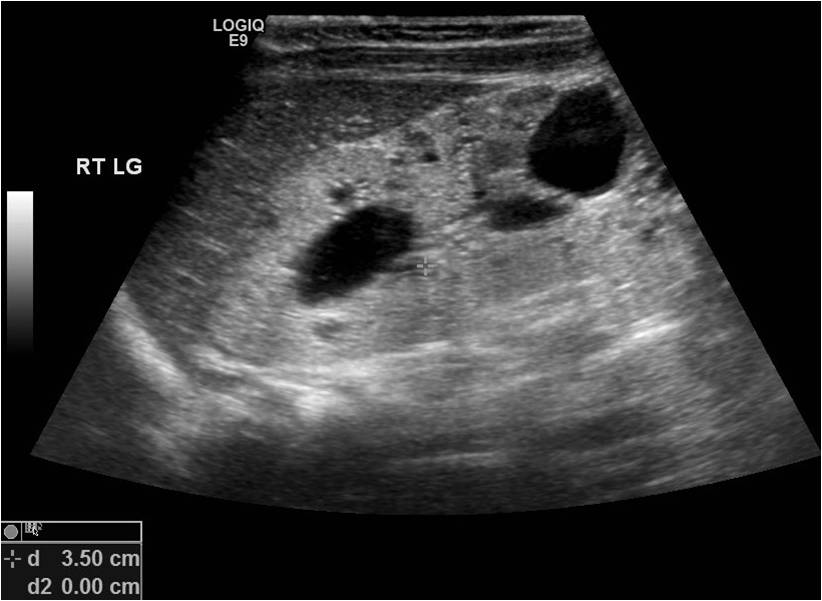
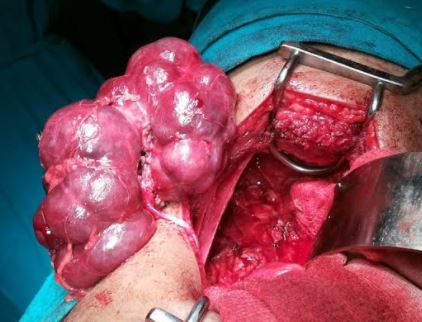
[1]
Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney international. 2009 Jul:76(2):149-68. doi: 10.1038/ki.2009.128. Epub 2009 May 20
[PubMed PMID: 19455193]
[2]
Lentine KL, Xiao H, Machnicki G, Gheorghian A, Schnitzler MA. Renal function and healthcare costs in patients with polycystic kidney disease. Clinical journal of the American Society of Nephrology : CJASN. 2010 Aug:5(8):1471-9. doi: 10.2215/CJN.00780110. Epub 2010 Jun 10
[PubMed PMID: 20538839]
[3]
Akoh JA. Current management of autosomal dominant polycystic kidney disease. World journal of nephrology. 2015 Sep 6:4(4):468-79. doi: 10.5527/wjn.v4.i4.468. Epub
[PubMed PMID: 26380198]
[4]
Guay-Woodford LM, Galliani CA, Musulman-Mroczek E, Spear GS, Guillot AP, Bernstein J. Diffuse renal cystic disease in children: morphologic and genetic correlations. Pediatric nephrology (Berlin, Germany). 1998 Apr:12(3):173-82
[PubMed PMID: 9630032]
[5]
Truong LD, Choi YJ, Shen SS, Ayala G, Amato R, Krishnan B. Renal cystic neoplasms and renal neoplasms associated with cystic renal diseases: pathogenetic and molecular links. Advances in anatomic pathology. 2003 May:10(3):135-59
[PubMed PMID: 12717117]
Level 3 (low-level) evidence
[6]
Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Advances in anatomic pathology. 2006 Jan:13(1):26-56
[PubMed PMID: 16462154]
Level 3 (low-level) evidence
[7]
Bergmann C, Senderek J, Küpper F, Schneider F, Dornia C, Windelen E, Eggermann T, Rudnik-Schöneborn S, Kirfel J, Furu L, Onuchic LF, Rossetti S, Harris PC, Somlo S, Guay-Woodford L, Germino GG, Moser M, Büttner R, Zerres K. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Human mutation. 2004 May:23(5):453-63
[PubMed PMID: 15108277]
[8]
Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. The New England journal of medicine. 2008 Oct 2:359(14):1477-85. doi: 10.1056/NEJMcp0804458. Epub
[PubMed PMID: 18832246]
[9]
Traubici J, Daneman A. High-resolution renal sonography in children with autosomal recessive polycystic kidney disease. AJR. American journal of roentgenology. 2005 May:184(5):1630-3
[PubMed PMID: 15855129]
[10]
Zhang Q, Taulman PD, Yoder BK. Cystic kidney diseases: all roads lead to the cilium. Physiology (Bethesda, Md.). 2004 Aug:19():225-30
[PubMed PMID: 15304637]
[11]
Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nature reviews. Molecular cell biology. 2007 Nov:8(11):880-93
[PubMed PMID: 17955020]
[13]
Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature. 2011 Aug 10:476(7359):163-9. doi: 10.1038/nature10275. Epub 2011 Aug 10
[PubMed PMID: 21833082]
[14]
Li X, Wüthrich RP, Kistler AD, Rodriguez D, Kapoor S, Mei C. Blood Pressure Control for Polycystic Kidney Disease. Polycystic Kidney Disease. 2015 Nov:():
[PubMed PMID: 27512778]
[15]
Martinez JR, Grantham JJ. Polycystic kidney disease: etiology, pathogenesis, and treatment. Disease-a-month : DM. 1995 Nov:41(11):693-765
[PubMed PMID: 7587886]
[16]
Richards WG, Sweeney WE, Yoder BK, Wilkinson JE, Woychik RP, Avner ED. Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. The Journal of clinical investigation. 1998 Mar 1:101(5):935-9
[PubMed PMID: 9486961]
[17]
Kamath BM, Piccoli DA. Heritable disorders of the bile ducts. Gastroenterology clinics of North America. 2003 Sep:32(3):857-75, vi
[PubMed PMID: 14562578]
[18]
Chapman AB. Approaches to testing new treatments in autosomal dominant polycystic kidney disease: insights from the CRISP and HALT-PKD studies. Clinical journal of the American Society of Nephrology : CJASN. 2008 Jul:3(4):1197-204. doi: 10.2215/CJN.00060108. Epub 2008 Jun 25
[PubMed PMID: 18579674]
[19]
Grantham JJ, Torres VE, Chapman AB, Guay-Woodford LM, Bae KT, King BF Jr, Wetzel LH, Baumgarten DA, Kenney PJ, Harris PC, Klahr S, Bennett WM, Hirschman GN, Meyers CM, Zhang X, Zhu F, Miller JP, CRISP Investigators. Volume progression in polycystic kidney disease. The New England journal of medicine. 2006 May 18:354(20):2122-30
[PubMed PMID: 16707749]
[20]
Chapman AB, Guay-Woodford LM, Grantham JJ, Torres VE, Bae KT, Baumgarten DA, Kenney PJ, King BF Jr, Glockner JF, Wetzel LH, Brummer ME, O'Neill WC, Robbin ML, Bennett WM, Klahr S, Hirschman GH, Kimmel PL, Thompson PA, Miller JP, Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney international. 2003 Sep:64(3):1035-45
[PubMed PMID: 12911554]
[21]
Gleason DC, McAlister WH, Kissane J. Cystic disease of the kidneys in children. The American journal of roentgenology, radium therapy, and nuclear medicine. 1967 May:100(1):135-46
[PubMed PMID: 4960727]
[22]
Sweeney WE Jr, Avner ED. Diagnosis and management of childhood polycystic kidney disease. Pediatric nephrology (Berlin, Germany). 2011 May:26(5):675-92. doi: 10.1007/s00467-010-1656-1. Epub 2010 Oct 29
[PubMed PMID: 21046169]
[23]
Chilton SJ, Cremin BJ. The spectrum of polycystic disease in children. Pediatric radiology. 1981:11(1):9-15
[PubMed PMID: 7254933]
[24]
Levine E, Hartman DS, Meilstrup JW, Van Slyke MA, Edgar KA, Barth JC. Current concepts and controversies in imaging of renal cystic diseases. The Urologic clinics of North America. 1997 Aug:24(3):523-43
[PubMed PMID: 9275977]
[25]
Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics. 2014 Sep:134(3):e833-45. doi: 10.1542/peds.2013-3646. Epub 2014 Aug 11
[PubMed PMID: 25113295]
[26]
Sweeney WE Jr, Avner ED. Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy. Pediatric research. 2014 Jan:75(1-2):148-57. doi: 10.1038/pr.2013.191. Epub 2013 Oct 31
[PubMed PMID: 24336431]
[27]
Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatric nephrology (Berlin, Germany). 1997 Jun:11(3):302-6
[PubMed PMID: 9203177]
[28]
Ruggenenti P, Remuzzi A, Ondei P, Fasolini G, Antiga L, Ene-Iordache B, Remuzzi G, Epstein FH. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney international. 2005 Jul:68(1):206-16
[PubMed PMID: 15954910]
[29]
Higashihara E, Torres VE, Chapman AB, Grantham JJ, Bae K, Watnick TJ, Horie S, Nutahara K, Ouyang J, Krasa HB, Czerwiec FS, TEMPOFormula and 156-05-002 Study Investigators. Tolvaptan in autosomal dominant polycystic kidney disease: three years' experience. Clinical journal of the American Society of Nephrology : CJASN. 2011 Oct:6(10):2499-507. doi: 10.2215/CJN.03530411. Epub 2011 Sep 8
[PubMed PMID: 21903984]
[30]
Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Perrone RD, Koch G, Ouyang J, McQuade RD, Blais JD, Czerwiec FS, Sergeyeva O, REPRISE Trial Investigators. Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease. The New England journal of medicine. 2017 Nov 16:377(20):1930-1942. doi: 10.1056/NEJMoa1710030. Epub 2017 Nov 4
[PubMed PMID: 29105594]
[31]
Patel V, Chowdhury R, Igarashi P. Advances in the pathogenesis and treatment of polycystic kidney disease. Current opinion in nephrology and hypertension. 2009 Mar:18(2):99-106. doi: 10.1097/MNH.0b013e3283262ab0. Epub
[PubMed PMID: 19430332]
Level 3 (low-level) evidence
[32]
Cowley BD Jr, Gudapaty S, Kraybill AL, Barash BD, Harding MA, Calvet JP, Gattone VH 2nd. Autosomal-dominant polycystic kidney disease in the rat. Kidney international. 1993 Mar:43(3):522-34
[PubMed PMID: 8455352]
[33]
Gile RD, Cowley BD Jr, Gattone VH 2nd, O'Donnell MP, Swan SK, Grantham JJ. Effect of lovastatin on the development of polycystic kidney disease in the Han:SPRD rat. American journal of kidney diseases : the official journal of the National Kidney Foundation. 1995 Sep:26(3):501-7
[PubMed PMID: 7645559]
[34]
Telega G, Cronin D, Avner ED. New approaches to the autosomal recessive polycystic kidney disease patient with dual kidney-liver complications. Pediatric transplantation. 2013 Jun:17(4):328-35. doi: 10.1111/petr.12076. Epub 2013 Apr 17
[PubMed PMID: 23593929]
[35]
Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2001 Oct:38(4):777-84
[PubMed PMID: 11576881]
[36]
Mercado-Deane MG, Beeson JE, John SD. US of renal insufficiency in neonates. Radiographics : a review publication of the Radiological Society of North America, Inc. 2002 Nov-Dec:22(6):1429-38
[PubMed PMID: 12432113]